Description of analysis methods
Sequence analysis methods
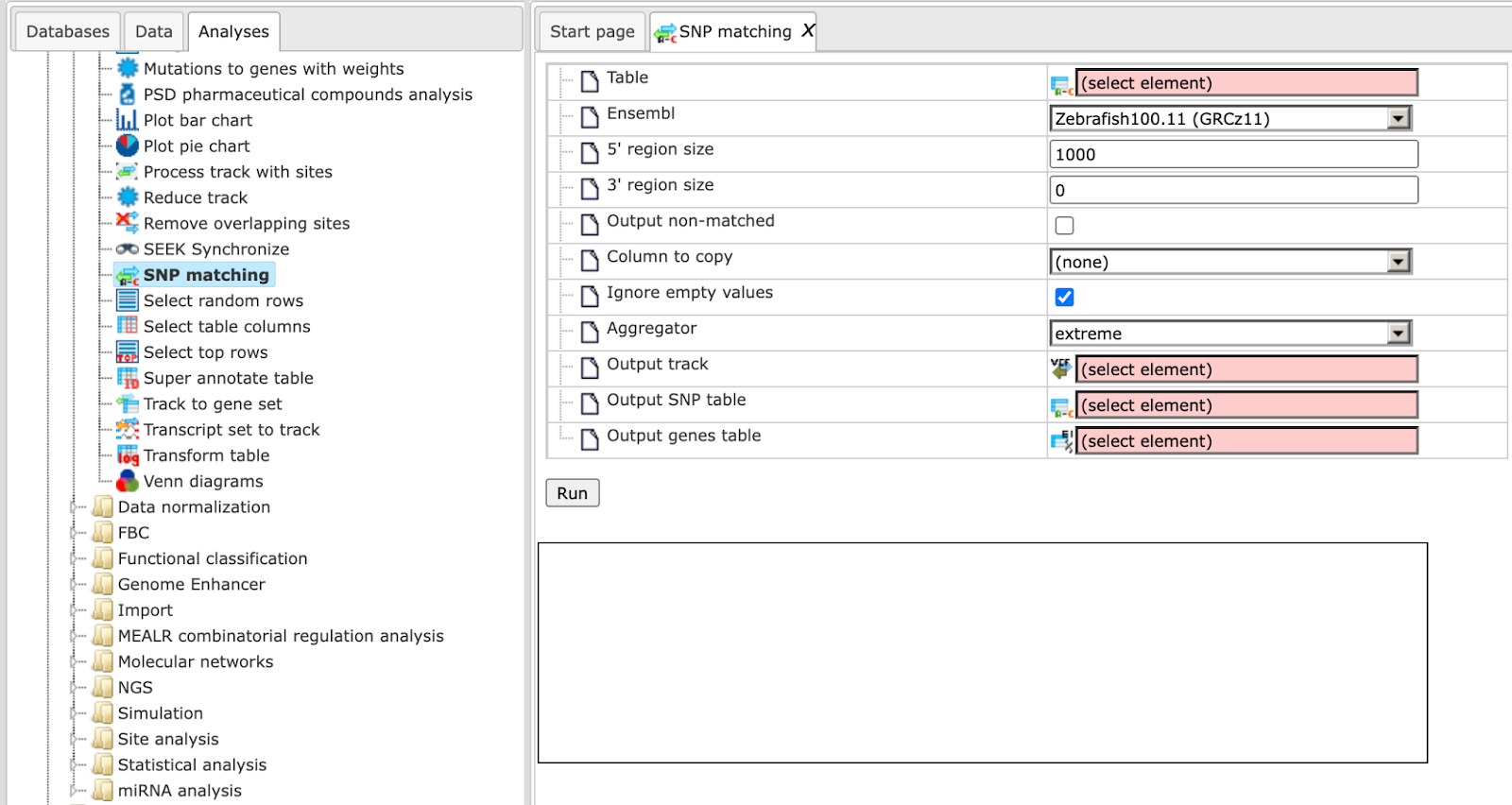
SNP matching
NGS DNA sequencing is a powerful analytical method to discover novel SNPs and detect known SNPs. The geneXplain platform provides a unique analysis method termed SNP matching. Using this method and an SNP table (derived after sequencing) as input, the corresponding SNP loci are mapped to Ensembl genes so that you can get an annotated SNP table as output.
By default the SNP matching tool looks as shown below:
Example:
An example data file to be used as an input table for SNP matching may look like this:
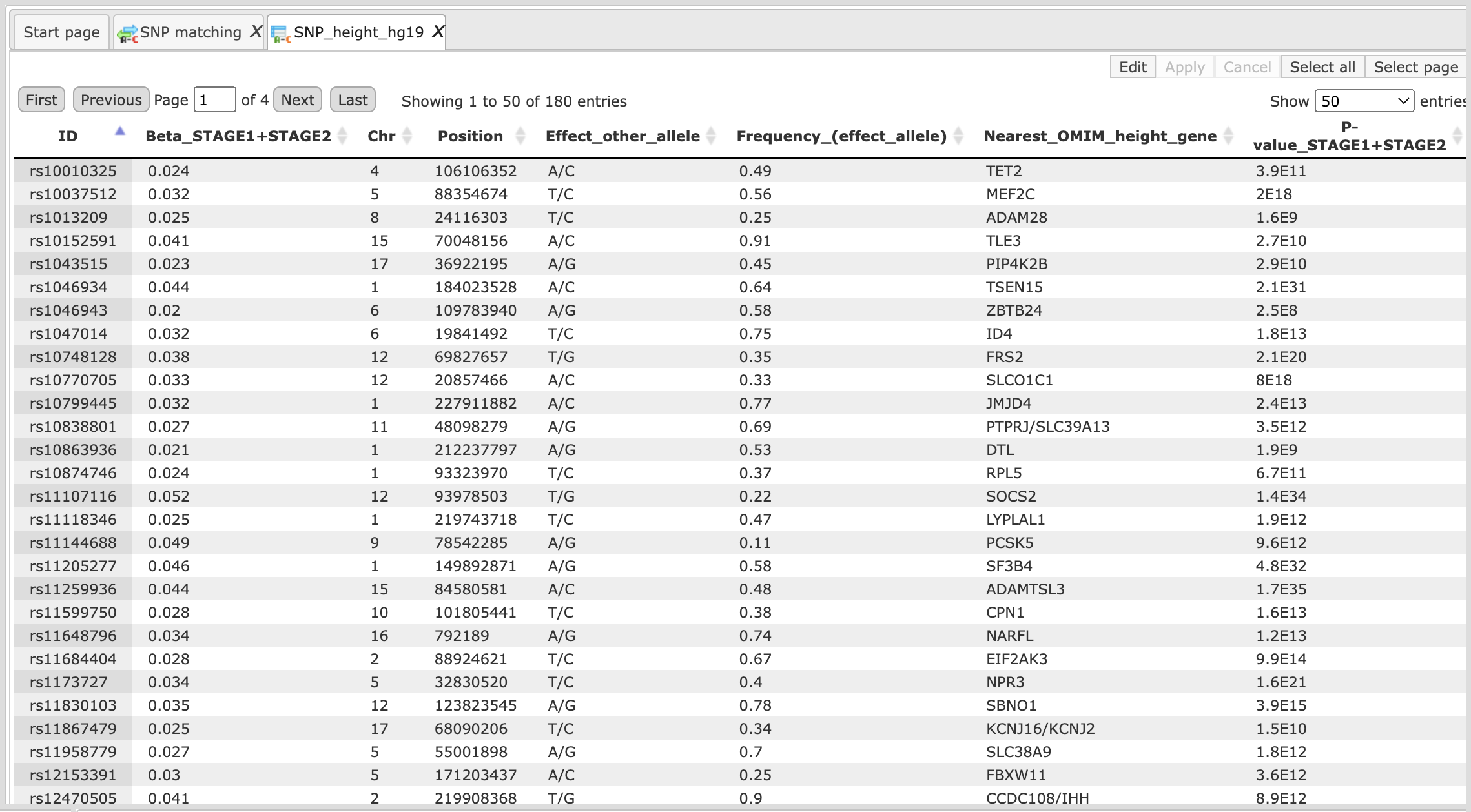
You can save this table in the repository and input the saved table in the SNP matching tool. The tool yields three output files as shown below:
Output table 1 (SNPs_annotated)
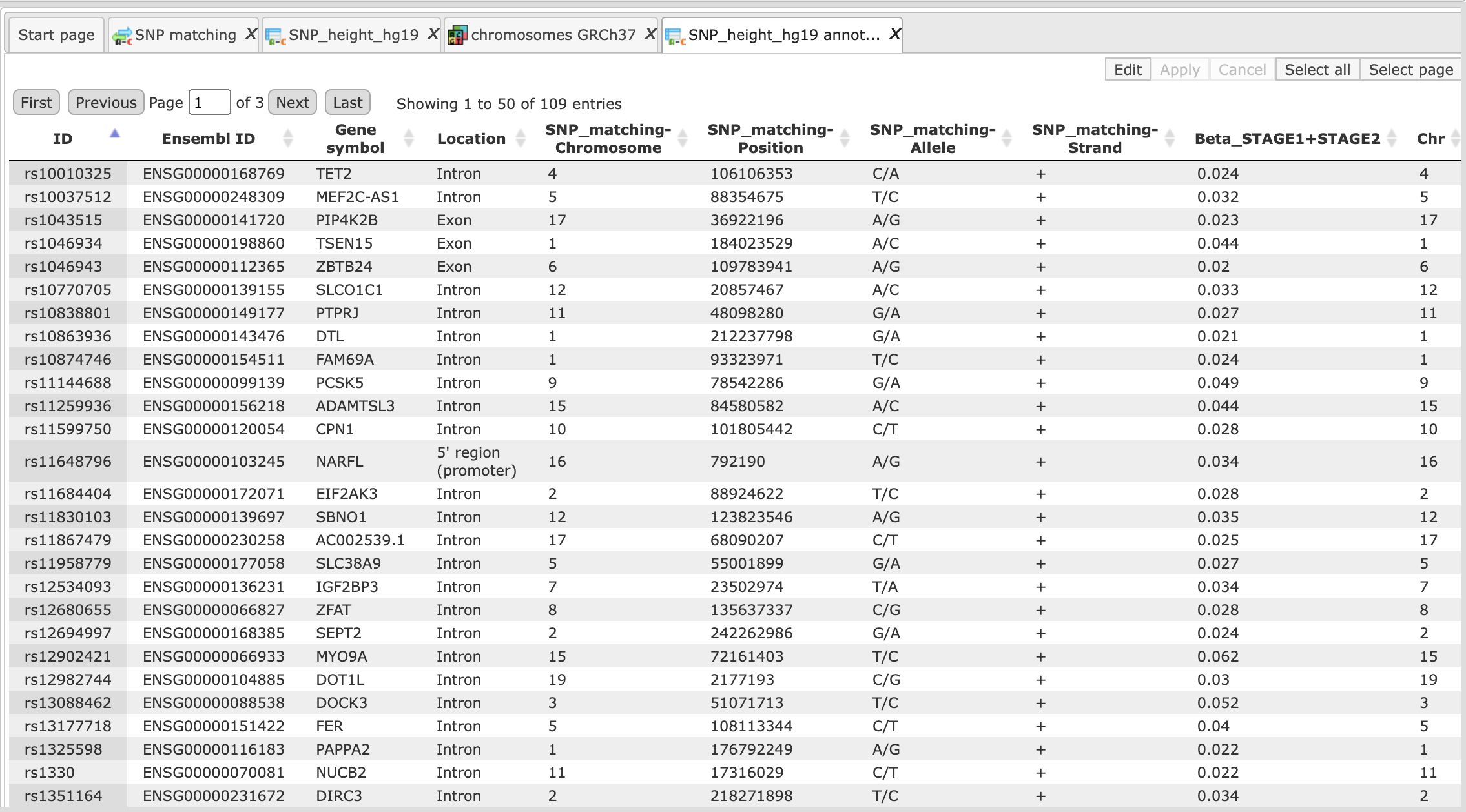
Output table 2 (SNPs_genes)
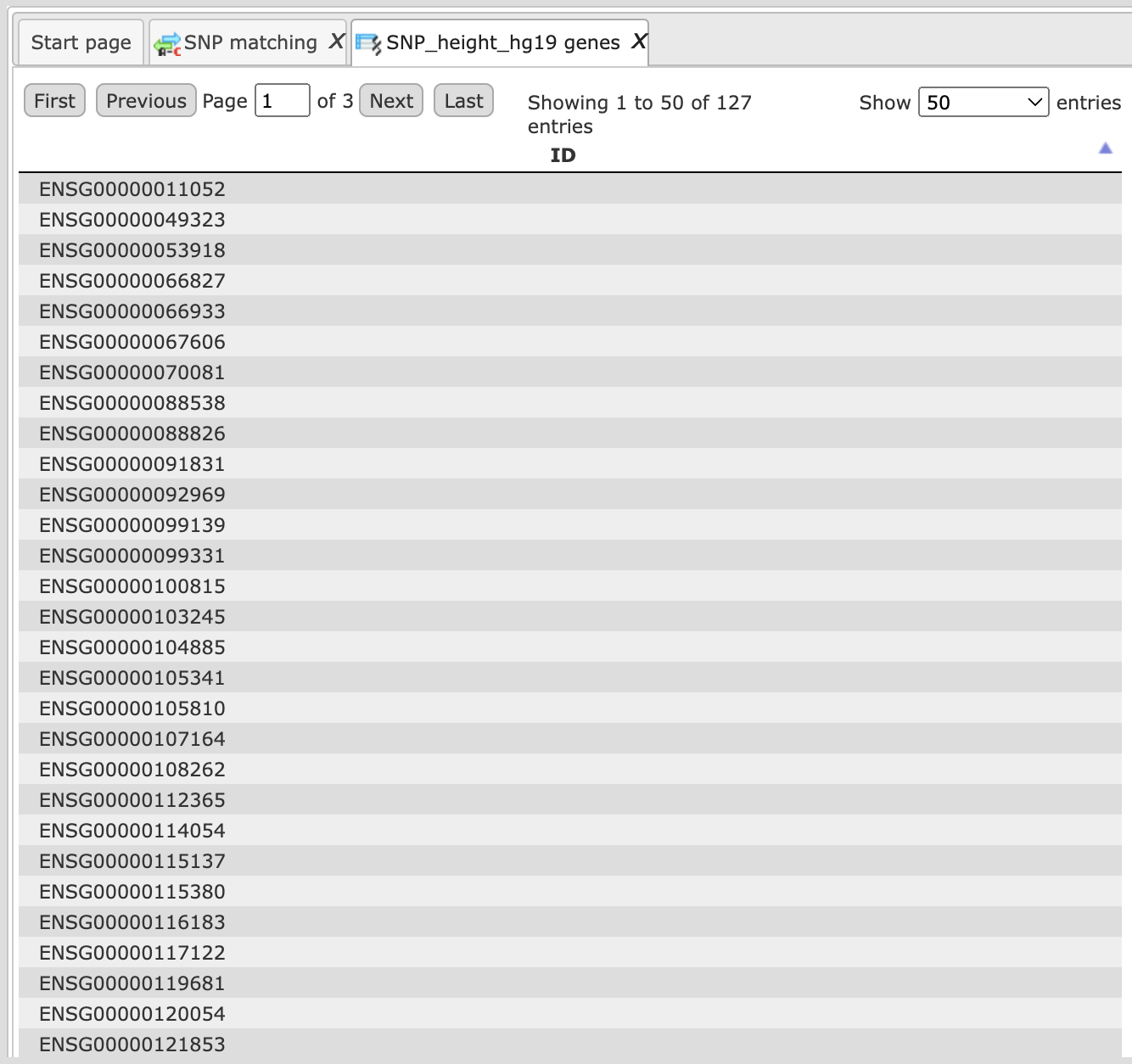
Output table 3 (track)
The output tables can be further used for any other analysis of the geneXplain platform.
Molecular networks
Goal: to search for important molecules in signal transduction cascade.
Input and output
Input: a set of genes/molecules to start analysis with. For instance, this can be a set of transcription factors, which may result from a promoter analysis or a set of ligands/receptors that trigger a certain (set of) pathway(s).
Two separate analyses are available:
Effector search for molecules downstream of the molecules in the input list
Regulator search for molecules upstream of the molecules in the input list
Output: a set of proteins or their encoding genes, which may play a key role in regulating (or being regulated by) a maximal number of start molecules.
Parameters
Molecules collection – Input the collection of molecules/genes
Weighting column (expert) – Column to replace weights in search graph
**Limit input size (expert) – Limit size of input list
Input size (expert) – Size of input list
Max radius – Maximal search radius
Score cutoff – Molecules with Score lower than specified will be excluded from the result
Search collection – Collection containing reactions
Custom search collection – Path to the custom search collection
Relation sign – Consider only specified type of relation chain between molecules.
Species – Species to which analysis should be confined
Calculate FDR – If true, analysis will calculate False Discovery Rate
FDR cutoff – Molecules with FDR higher than specified will be excluded from the result
Z-score cutoff – Molecules with Z-score lower than specified will be excluded from the result
Random seed – Seed for FDR random generator
Penalty (expert) – Penalty value for false positives
Decorators – Possibility to modify search collection with custom interaction sets
Normalize multi-forms (expert) – Normalize weights of multiple forms
Output name – Output name.
Alforithm description
In drug target search analysis one searches for signaling molecules and corresponding networks that can transmit a signal to or receive a signal from several of input molecules within a certain limit of reaction steps. A search starts from each molecule of an input set Vx and constructs the shortest paths to all nodes V of the complete network within a given maximal path cost R (i.e, the sum of the costs of all edges in the shortest path from a vertex in Vx to a vertex in V should be smaller than or equal to R). The search can be conducted in reverse direction of the edges leading to input molecules (upstream) or in the same direction (downstream).
The Specificity score is calculated for every molecule found according to:
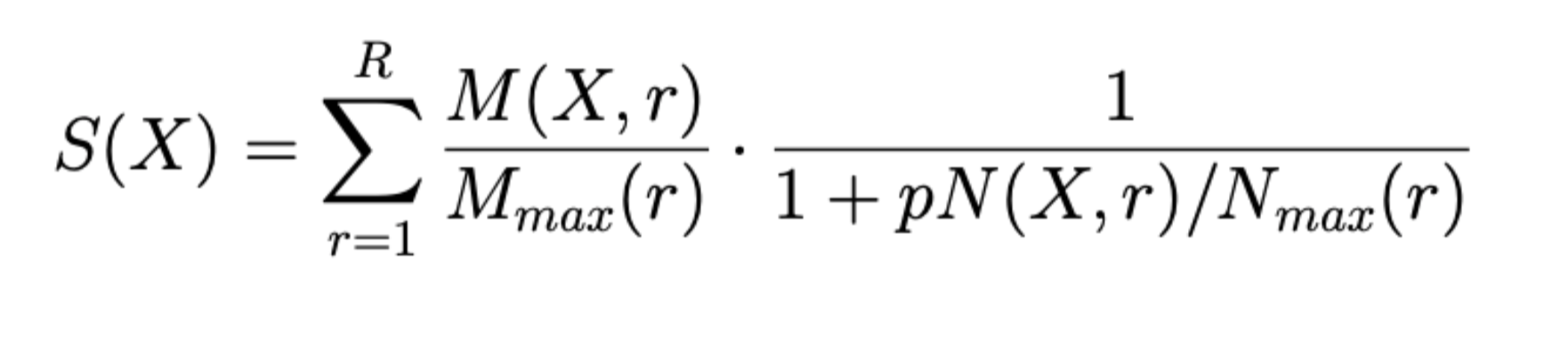
Where:
R — Max radius (input parameter)
p — Penalty (input parameter)
N(X,r) — total number of molecules reachable from key molecule X within the radius r.
Nmax(r) — maximal value of N(X,r) over all key molecules X found for this radius.
M(X,r) — sum of w(X) for all hits reachable from key molecule X within the radius r, where w(X) — weight of hit X. It equals to wb(X) if “Normalize multi-forms” is unchecked. Otherwise it’s wb(X)/I(X), where I(X) is the number of multiforms of X in the input set (not total number of multiforms in the database). In both cases wb(X) is the base weight of hit X. It equals the corresponding value in “Weighting column” or 1 if “Weighting column” is not specified.
Mmax(r) — maximal value of M(X,r) over all key molecules X found for this radius.
FDR
Each individual drug target molecule gets a p-value (FDR) assigned, which represents the probability to occupy the observed rank or higher ranks by random chance. It is estimated on-the-fly by random sampling. The ranking of the key nodes is defined by sorting them according to the Score above in descending order. It should be noted that the rank is defined by the ranks of the occurring scores, which means that more than one key node can share the same score value in some cases. Molecules which do not have any hits get assigned the last rank since the score is zero in this case.
Z-Score
In addition to the FDR, each drug target molecule gets a Z-Score
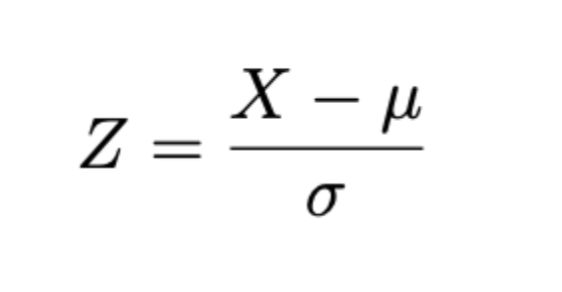
which measures the deviation of the observed rank X of the key node from the expected rank μ in random case, divided by the standard deviation. In this formula, the rank above distribution is assumed to comply the normal distribution. Key nodes with Z greater than 1.0 are considered significant.
Context algorithm
For the purpose of incorporating additional contextual knowledge, e.g. a certain disease which we know to be related to the anticipated analysis, we implemented a method which encodes this additional context information as modified edge costs in the signaling network. The context information has to be provided as a second gene set (context genes). The idea is based on attracting the drug target molecule search (e.g. the underlying Dijkstra algorithm for shortest paths) towards context genes by decreasing the costs of those edges that are close to the context genes. It features two major aspects:
Attraction (”gravity”) of the shortest-paths towards context genes C
Distribution of the attraction power to an extended surrounding area around C in order to prefer shortest paths close to context genes in case there is no path possible that goes through the context gene directly. (”gravity range”).
Result columns
ID — key molecule identifier in respective database
Key molecule name — molecule title
Reached from set — number of molecules from input set, that were reached from key molecule within the distance given
Reachable total — total number of molecules, that can be reached from key molecule within the distance given
Score — specificity score value calculated as described above
FDR — p-value, which represents the probability to occupy the observed rank or higher ranks by random chance
Z-Score — z-score, according the equation above
Hits — identifiers of molecules from input set, that were reached from key molecule within the distance given
Hits names — titles of molecules from input set, that were reached from key molecule within the distance given
References
Kel, A., Voss, N., Jauregui, R., Kel-Margoulis, O. and Wingender, E.: Beyond microarrays: Find key transcription factors controlling signal transduction pathways BMC Bioinformatics 7(Suppl. 2), S13 (2006).
Site search on gene set
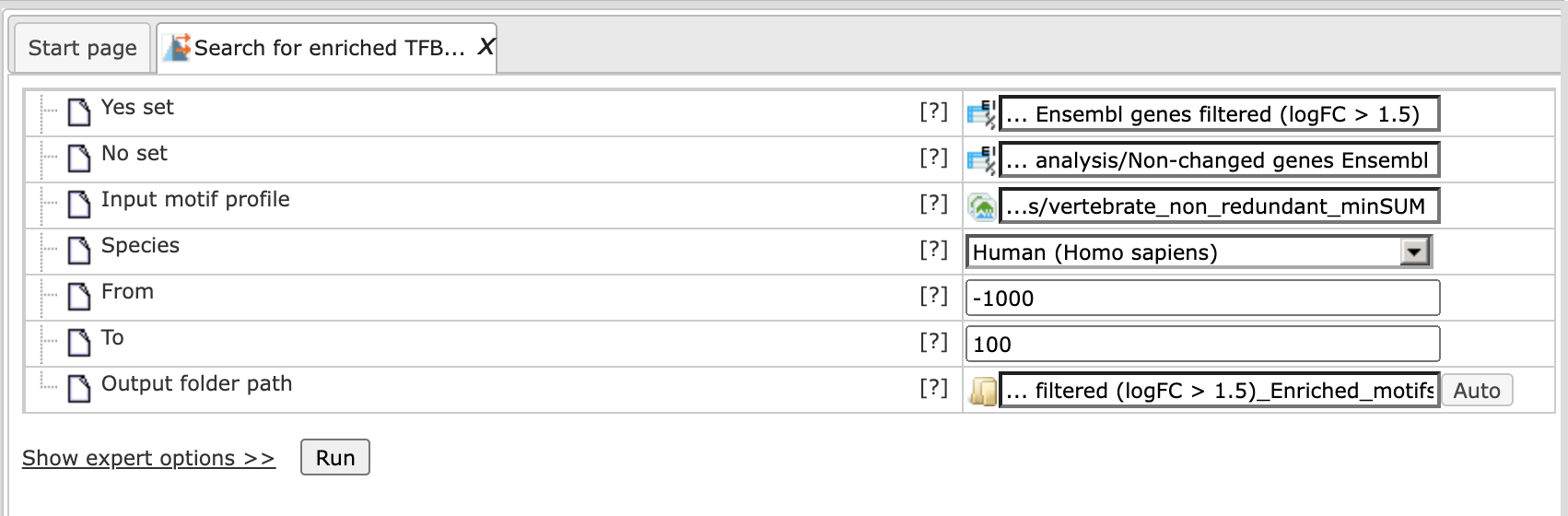
This feature provides you with an option to search for putative transcription factor binding sites (TFBS) in a set of genes. As input for the analysis you are supposed to indicate two gene sets, Yes (e.g. differentially expressed in an experiment, test set) and No (set of background genes, control set) as well as positional range relative to the TSS and a collection of predefined weight matrices with a particular threshold (profile).
The initial form of this analysis looks as it is shown below:
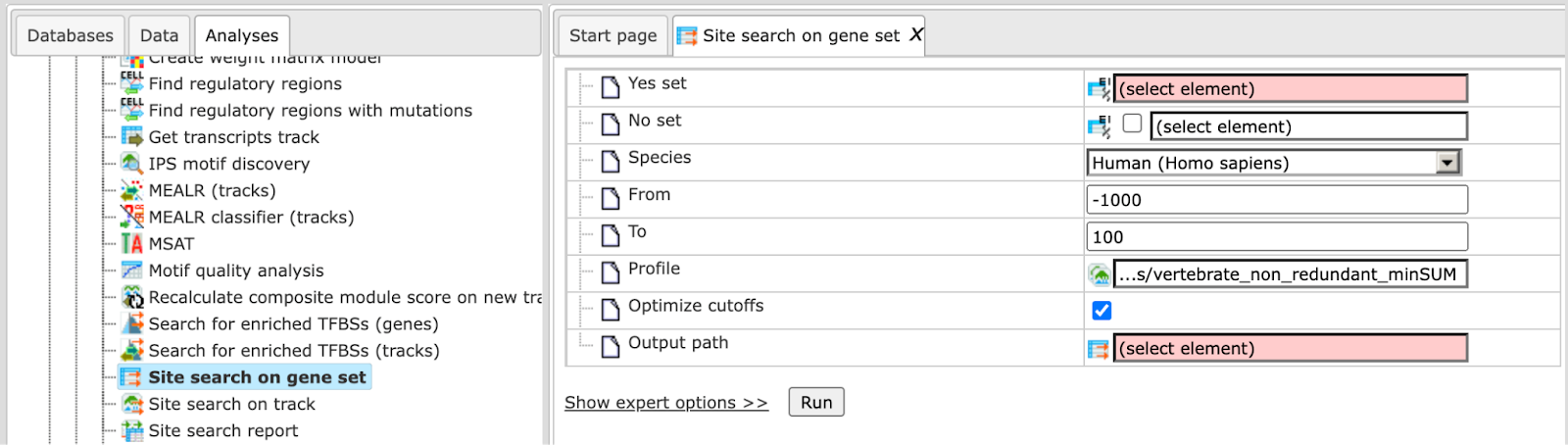
To perform this analysis you have to input two datasets:
Yes set: This is the set of genes that you want to analyze, for example these can be genes the expression of which has changed in an experiment (test set). Tables with Ensembl gene IDs can be used as input in this column. In case you have a file with different identifiers, you can first use the Convert table function in the Data manipulation folder.
No set: This is the set of background genes (control set). This again should be a gene table with Ensembl gene IDs as input.
These two datasets might be taken from the output tables of previous analyses; see, e.g., Detect differentially expressed genes as described previously.
Species: A pull down menu allows you to select the biological species according to the species of your Yes and No gene sets. Currently, the analysis can be done for human, mouse and rat genes.
From: You can indicate the gene region where the search for putative TFBS should be done. Here, you enter the 5’ border of the region, relative to the transcription start site (TSS) as it is annotated in the current version of Ensembl.
To: Here, you enter the 3’ border of the region relative to the TSS.
Profile: This is a predefined set of positional weight matrices with a particular threshold. By default, the TRANSFAC® profile vertebrate_non_redundant_minSUM is applied. You can use other available TRANSFAC® profiles. Alternatively, you can apply profiles in the GTRD database. Currently, there are three profiles included in the GTRD database; they depend on the threshold for matrices: strong threshold (with an Individual Probability Score (IPS) higher than 6), moderate threshold (with IPS higher than 5), and weak threshold (with IPS higher than 4).
You also have the option to import matrices and profiles into the geneXplain platform and use them for your further analysis.
To perform site search following steps are recommended:
Step 1. Input Yes set from the tree
Step 2. Input pre-saved No set. You can drag-and-drop as usual. Here, the set of human housekeeping genes from the Example folder is used as input No set, highlighted blue on the screenshot below:
Step 3. Select the species and the promoter length. By default the promoter
length is set as -1000 to +100 relative to the TSS. In this example, the range
-500 to 100 is selected.
Step 4. Select the TRANSFAC® or GTRD profile from the pre-saved profiles in the tool. In this example the default TRANSFAC® profile vertebrate_non_redundant_minSUM is used.
Step 5. Identify the output path. Define where the folder with the results should be located in your project tree. You can do so by clicking on the pink field “select element” in the field Output path, and a new window will be opened, where you can select the location of the results folder and define its name.
Press [Run].
The analysis will start as shown below:
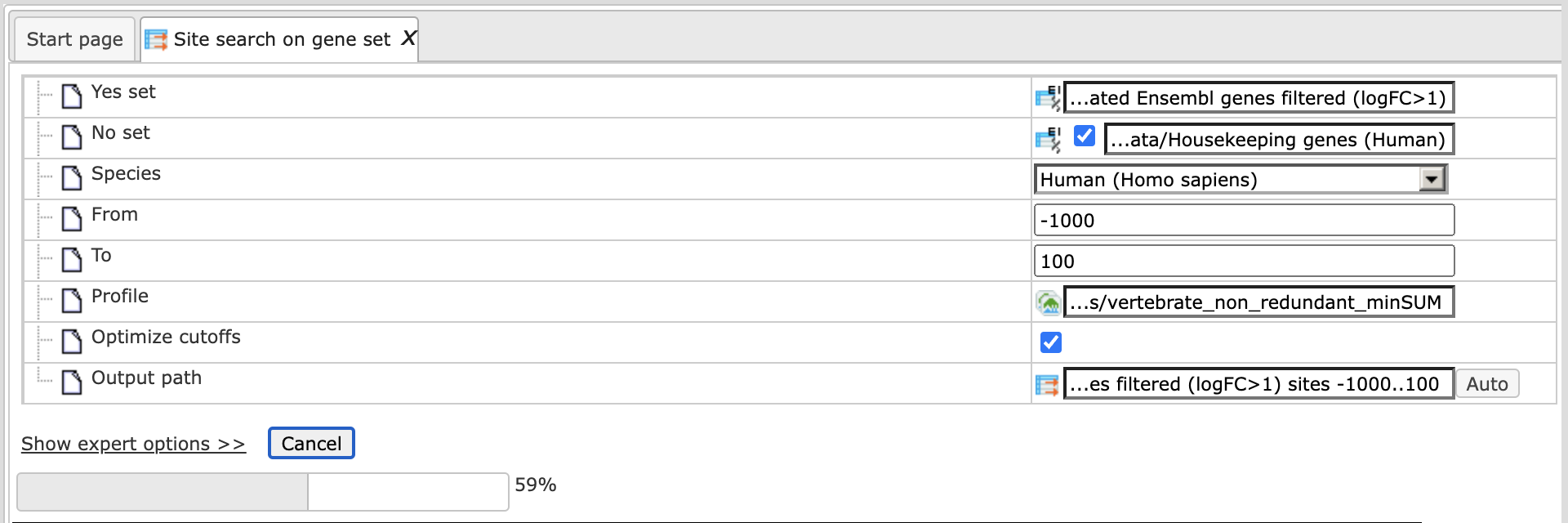
Wait until the analysis is complete as shown by the progress bar. The output of Site search on gene set contains one table and six tracks:
summary table ( ), yes promoters (
), yes promoters ( ), no promoters (), yes sites (), no sites (), yes sites optimized () and no sites optimized ()
), no promoters (), yes sites (), no sites (), yes sites optimized () and no sites optimized ()
The summary table is automatically opened in a new tab of the geneXplain platform when the analysis is completed. An example of the summary table is shown below:
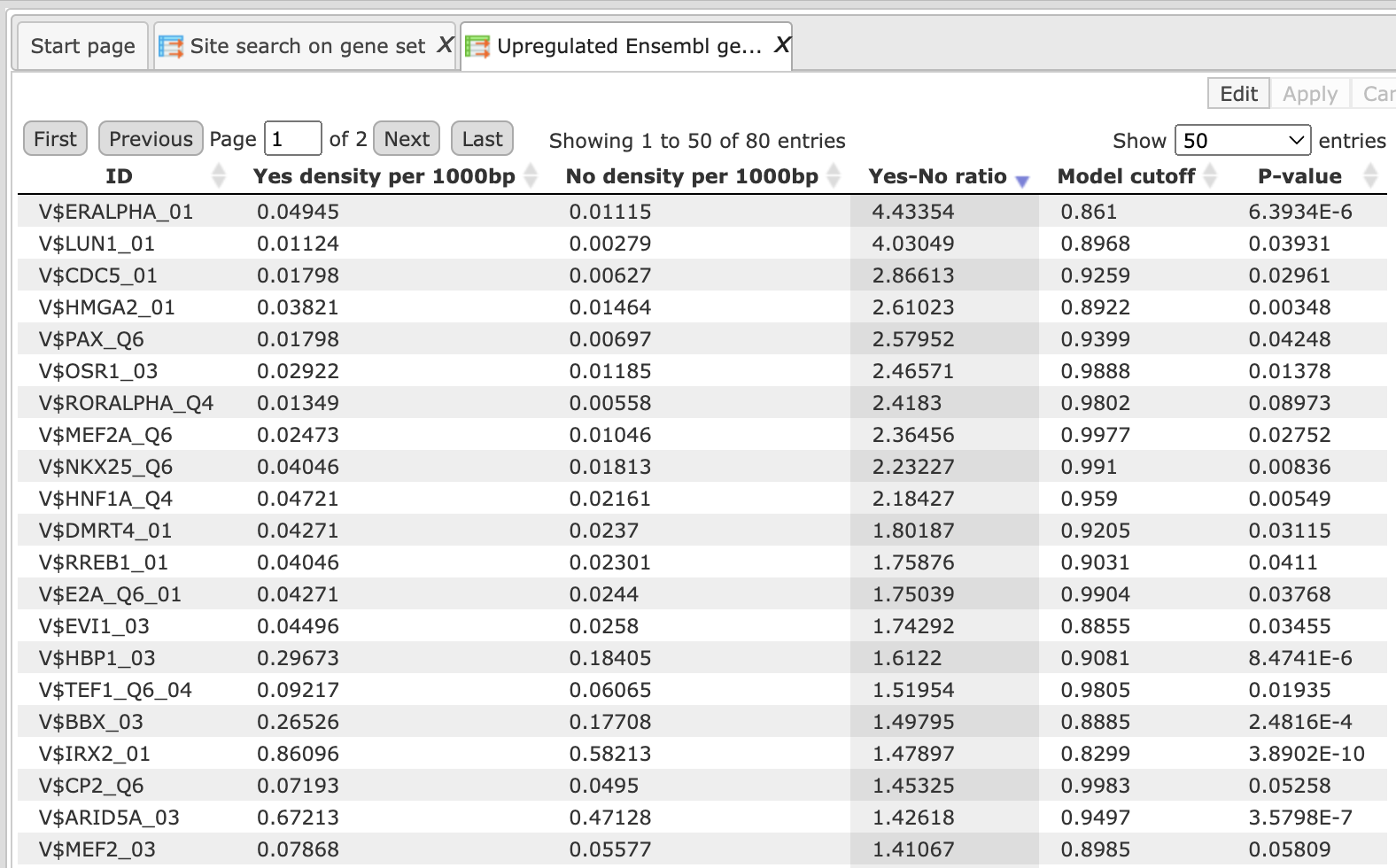
Each row summarizes the information for one PWM. For each selected matrix, the columns Yes density per 1000bp and No density per 1000bp show the number of matches normalized per 1000 bp length for the sequences in the input Yes set and input No set, respectively. The Column Yes-No ratio is the ratio of the first two columns. Only matrices with a Yes-No ratio higher than 1 are included in the summary table. The higher the Yes-No ratio, the higher is the enrichment of matches for the respective matrix in the Yes set. The matrix cutoff values as they are calculated by the program at the optimization step are shown in the column Model cutoff, and the last column shows the p-value of the corresponding event.
Tracks Yes promoters and No promoters
These two files resulting from the “Site search on gene set” analysis represent
promoters of the corresponding set, as a track ( ). The track files might be used for site visualization or for further analyses, e.g. Construct Composite Modules, Construct IPS CisModule.
). The track files might be used for site visualization or for further analyses, e.g. Construct Composite Modules, Construct IPS CisModule.
The track file Yes promoters when opened in the work space is shown below. The track file No promoters has a similar structure.
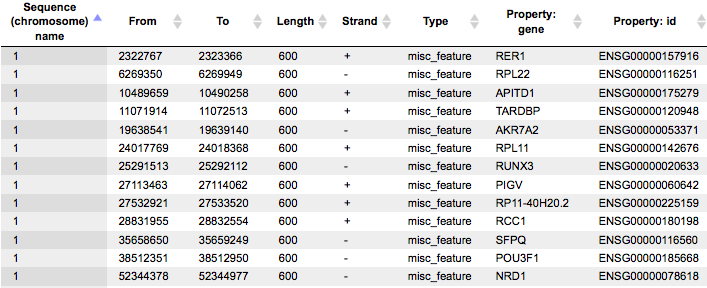
This table lists the positions of the promoter areas selected for the analysis on particular chromosomes, as shown in the columns From and To. The column Strand shows the strand on which each particular promoter is located. This track can be dragged and dropped on a particular chromosome opened in the genome browser to visualize the localizations of the promoters.
Yes (No) sites optimized track: The file Yes sites visualizes those putative sites that are over-represented in the promoters of the Yes set versus the No set as they are located in the promoters of the Yes set. Putative TFBSs are shown as a track
Scores of the putative sites are optimized by the algorithm.
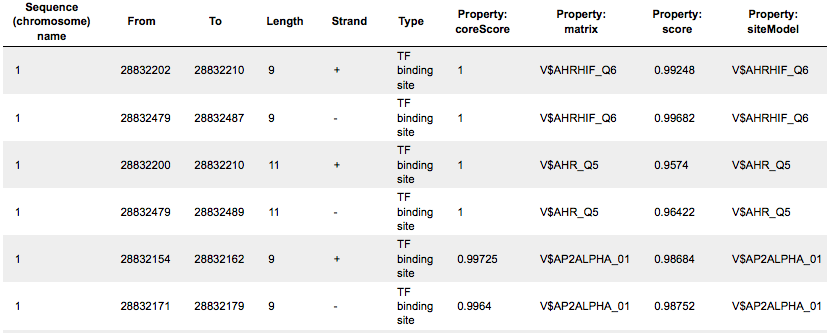
This track is a list of all putative TFBS found in one analysis. Each row presents details for each individual match for every PWM. The columns Sequence (chromosome) name, From, To, Length and Strand show, correspondingly, genomic location of the match including chromosome number, start and end positions, strand and length of the match. The column Type contains information about the type of the elements, in this case all matches are considered as “TF binding site”. Further columns keep information about PWM producing each match (column Property: matrix) as well as score for the whole matrix (column Property: score). The column Property: siteModel contains the identifier for the corresponding site model, which is the matrix together with a cutoff applied (and in the example shown is identical to the matrix identifier).
Yes (No) sites tracks are very similar in structure. The major difference is that these tracks include putative binding sites before the cutoff optimization, and thus they contain more sites.
These track files can be used as an input for other functions, for example sites can be visualized on chromosomes. For visualization details please refer to the tip in the Section sites visualization.
Visualization of TFBS for individual genes and for individual matrices
Having the summary table opened in the work space, you can select different rows and apply a couple of different functions with the help of the buttons on the top menu bar. These five buttons are in the top middle in the figure below.
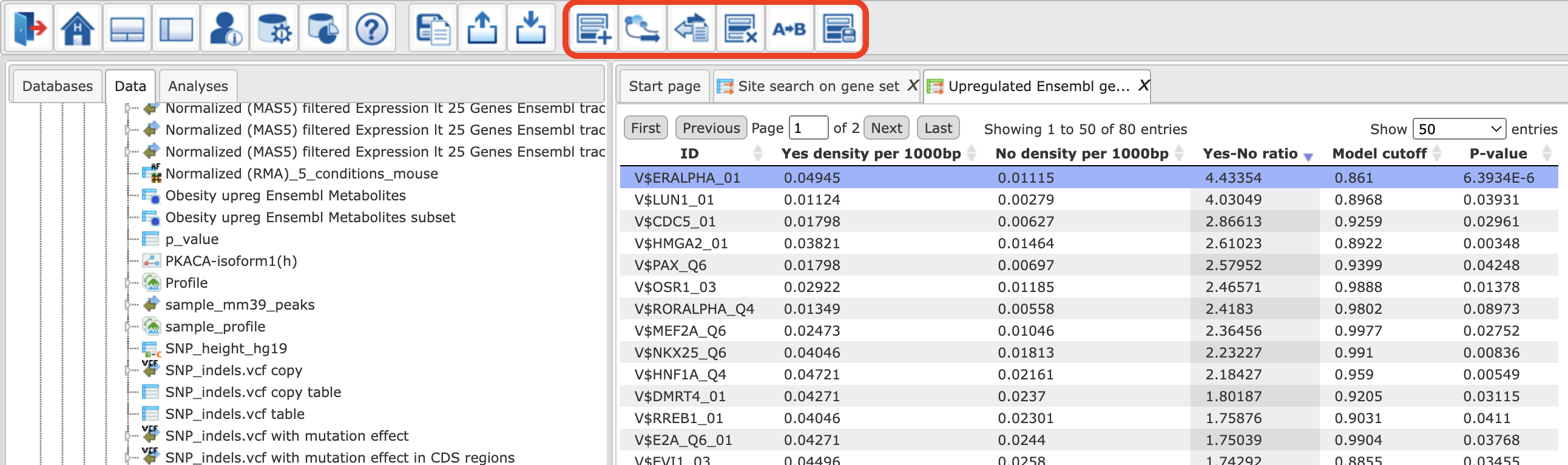
To get a visualization of TFBS for individual genes, the button  should be applied on the selected matrices. After click on this button, two new files will be saved in the tree area,
a table
should be applied on the selected matrices. After click on this button, two new files will be saved in the tree area,
a table  and a track , as shown below. The table is automatically opened in the Work Space.
and a track , as shown below. The table is automatically opened in the Work Space.
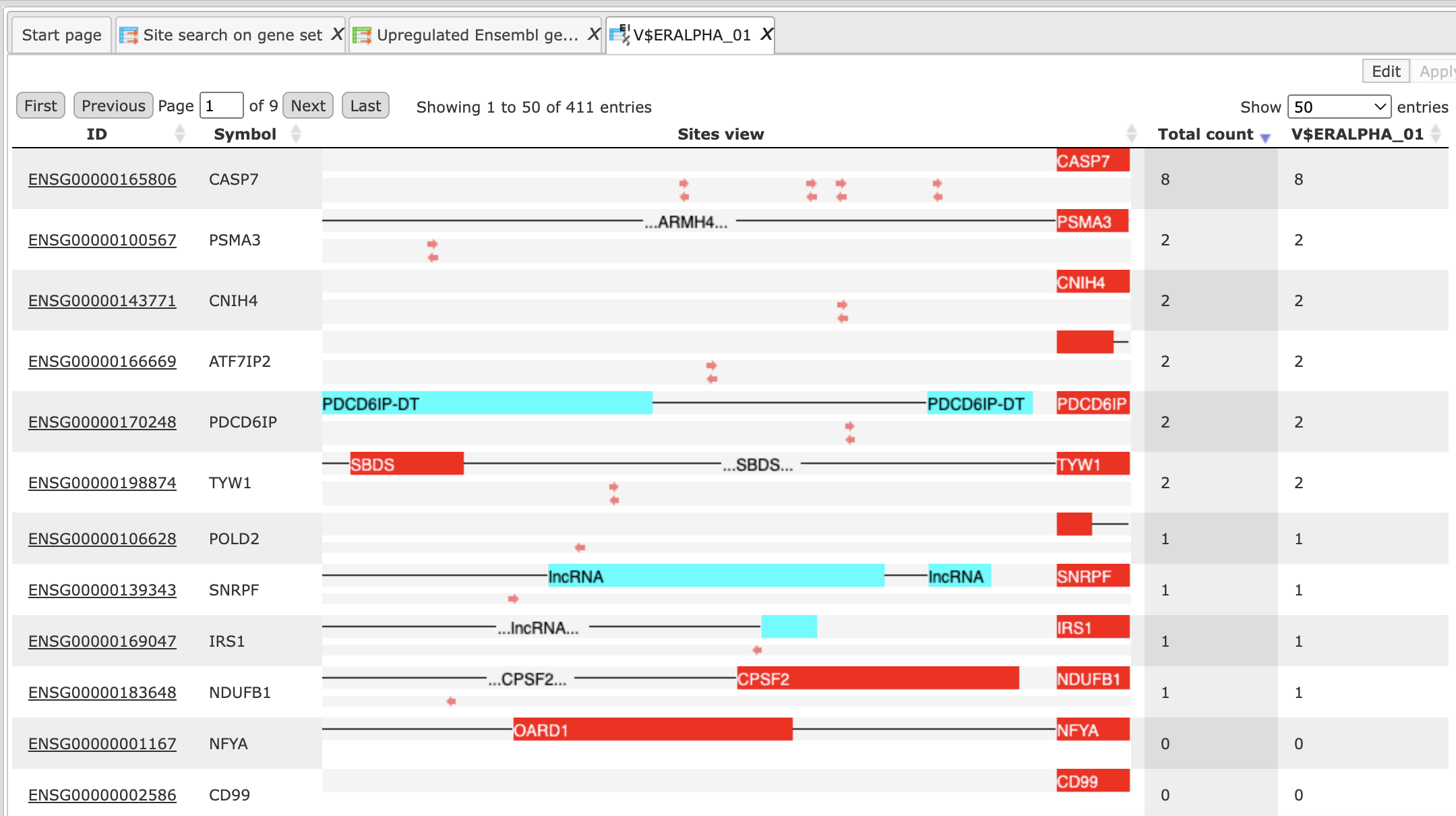
Let’s consider this table in more detail. Each row corresponds to one individual gene.
The column ID presents the Ensembl ID for each gene, and the gene symbol is shown in the column Symbol. The column Sites view shows a schematic representation for each gene, where blue bars correspond to gene starts and coding regions, and TFBSs for different matrices are shown by arrows of different colors. The column Total count shows the number of TFBSs for all matrices together in the promoter of each particular gene. The next columns are named as matrices in the summary table and represent the number of TFBSs for each matrix in each particular gene.
In the picture above the table is sorted by the column Total count, and on the top we can see those genes that contain the highest total number of sites. This table can be sorted by different columns corresponding to individual matrices, and then on the top you will see those genes that contain the highest number of sites for the matrix in focus.
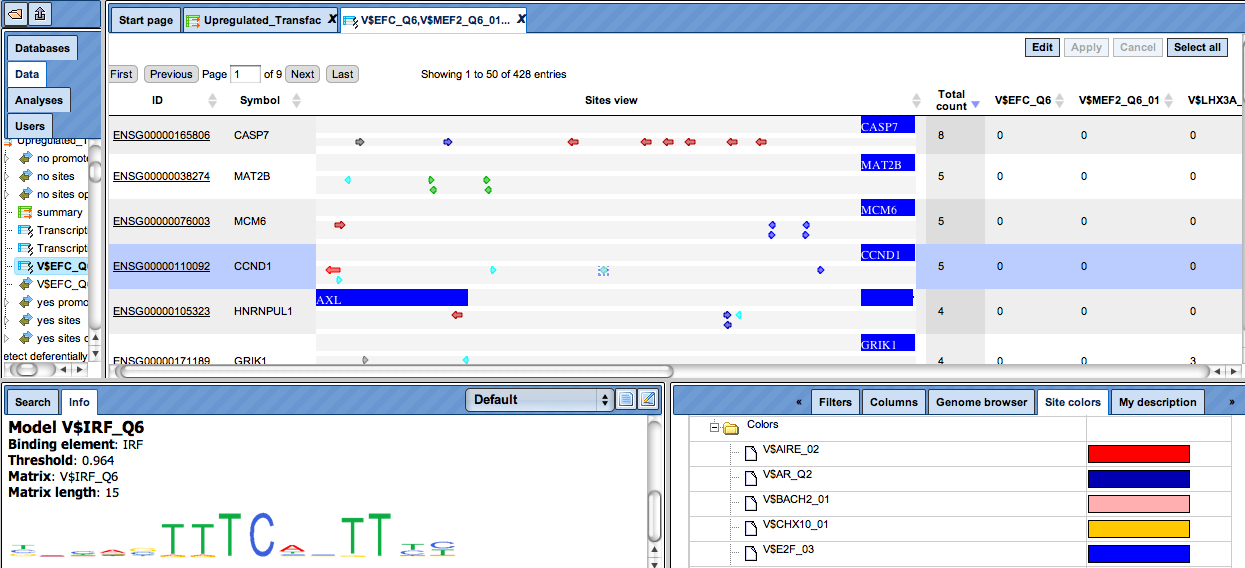
The TFBS color schema can be customized. For this, open the tab „Site colors“ in the Operation Field in the bottom right area of the tool (figure below). You can see the default colors for different matrices, and can adjust them by clicking on each color box.
This table can be exported in tab-separated format (txt) or comma-separated format (csv).
The second file, a track has the same structure as described above for other track files. For visualization details please refer to the tip in Section visualization of sites.
Creation of customized profiles
“Profile” is a term used for a collection of positional weight matrices (PWMs) with a particular threshold (or cutoff), also referred to as “site models”. The geneXplain platform provides an option to create profiles from a table of site models or from any gene table that contains some transcription factor genes. The newly created customized profiles can be further used for analyses of regulatory regions.
There are two possibilities to create a user-specific profile.
Create profile from gene table  This option supports you in creating a new profile from any gene table. The resulting profile contains site models linked to the genes encoding the corresponding transcription factors in the input gene set. This option can be accessed via the URL: https://platform.genexplain.com/bioumlweb/#de=analyses/Methods/Site%20analysis/Create%20profile%20from%20gene%20table
This option supports you in creating a new profile from any gene table. The resulting profile contains site models linked to the genes encoding the corresponding transcription factors in the input gene set. This option can be accessed via the URL: https://platform.genexplain.com/bioumlweb/#de=analyses/Methods/Site%20analysis/Create%20profile%20from%20gene%20table
Create profile from table  Here you can create a profile from a table of matrices. This option can be accessed via the URL:
https://platform.genexplain.com/bioumlweb/#de=analyses/Methods/Site%20analysis/Create%20profile%20from%20site%20model%20table
Here you can create a profile from a table of matrices. This option can be accessed via the URL:
https://platform.genexplain.com/bioumlweb/#de=analyses/Methods/Site%20analysis/Create%20profile%20from%20site%20model%20table
Create profile from gene table
This function creates a new profile from any gene table. The resulting profile contains site models linked to the transcription factors in the input gene set.
The input form when opened in the work space is shown below:
In the following, we will consider each of the input fields.
Gene set: Input any gene table here for which you intend to design a profile. The algorithm automatically identifies transcription factor genes in the input table and the matrices linked to these factors in the TRANSFAC® database. These matrices will be put into the newly created profile.
Further steps are demonstrated with the table of genes present here:
Species: Select the biological species corresponding to the input gene set from the drop-down menu.
Reference profile: Specify one of the TRANSFAC® profiles available. Cutoffs for the corresponding matrices will be copied from the selected reference TRANSFAC® profile into the newly created profile.
To select the reference profile click on the box of this field and a new window will be opened. Scroll towards the desired profile and click on it so that the name of the selected profile is displayed in the field “Name”. When the selection is done, press [Ok].
Please note that this analysis method works only for the TRANSFAC® profiles.
Output path: Specify the path to store the result and indicate the name for the new profile.
Having filled all the input fields, launch the analysis with the [Run] button.
After completion of the analysis the output profile is opened automatically as shown below:
Let us now have a look into the newly created profile.
Each row presents the information for one site model. In the column Name the
name of the site model is given which here is the same as for the matrix. In the
column Matrix name the name for the positional weight matrix is present. For
each site model, a cutoff adapted from the “Reference profile” is shown in the
column Cutoff.
According to the TRANSFAC® standard, the core part is specified for each matrix.
The core is represented by the 5 consecutive most conserved nucleotides. The
columns Core cutoff, Core start and Core length provide details about the core
of each matrix. In the last column the matrix logo is shown.
In the Tree Area, the newly created profile has the symbol  , the same symbol as for all other profiles, and is ready to use for the analysis of regulatory regions.
, the same symbol as for all other profiles, and is ready to use for the analysis of regulatory regions.
Suggestion when to use “Create profile from gene table”
If you plan to do a promoter analysis for differentially expressed genes from a particular experiment, you might be interested in creating a profile from a table of genes expressed under the same conditions. For example, you might take one of the tables resulting from the workflows “Detect differentially expressed genes…” as input gene table for profile creation.
Such a profile contains matrices for the transcription factors expressed under the same experimental conditions, and thus it might be reasonable to apply it for the promoter analysis of genes up-regulated or down-regulated in this experiment.
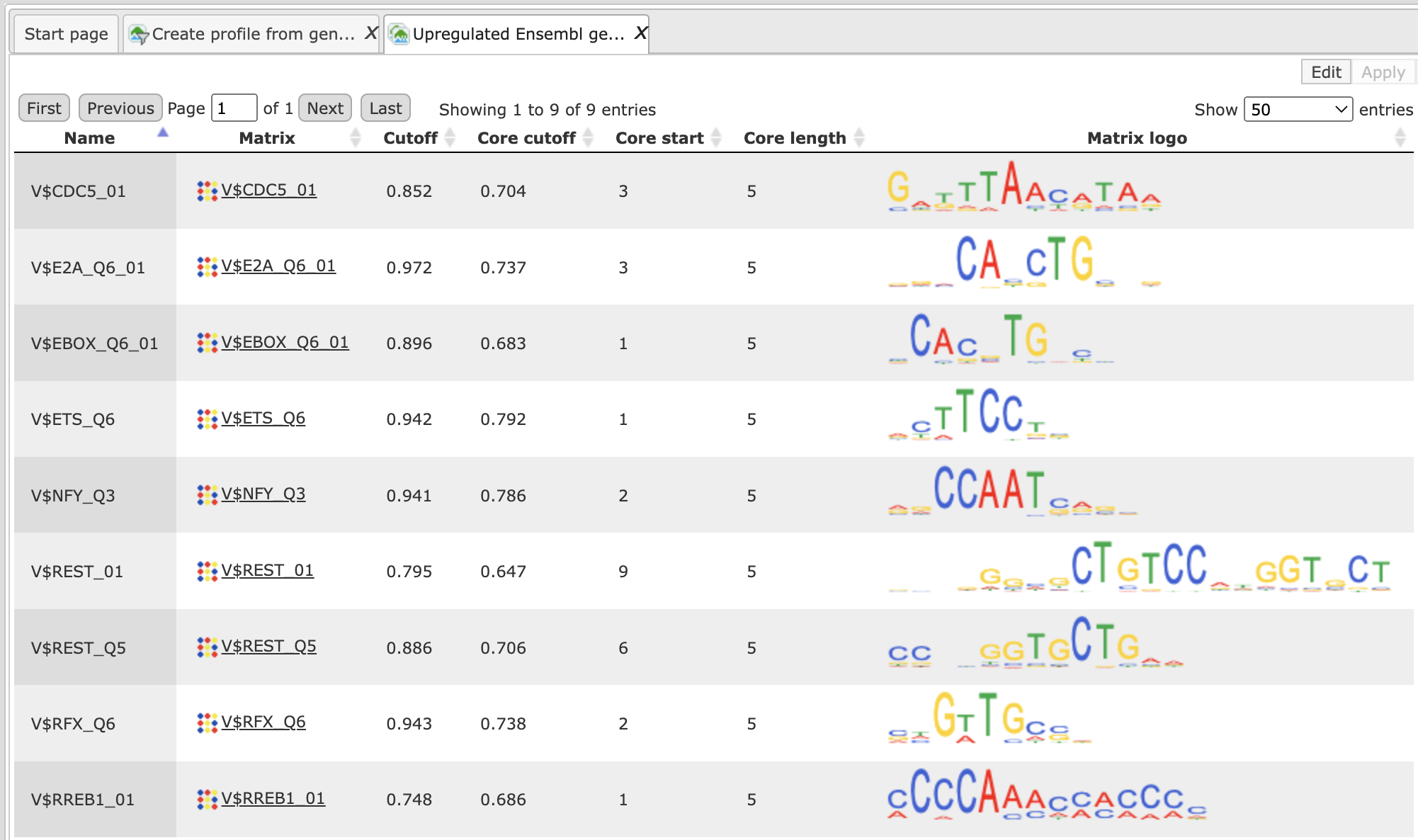
Create profile from site model table
This function creates a profile from the table of site models. For example, a new profile can be created from the summary table resulting from the workflow “Analyze promoters (TRANSFAC®)” or “Upstream analysis (TRANSFAC® and TRANSPATH®)”.
The analysis input form can be found in the Tree Area, under the Analyses tab, in the folder Methods/Site analysis. The opened input form is shown below.
Let us consider the individual input fields:
Input table: Input a table with matrices for which you intend to design a profile. The input table should contain TRANSFAC® matrix IDs as row names. Such a table can be the result of a site search analysis.
In the following, further steps are demonstrated with a table of site models from one of the examples. The example file can be accessed using the URL:
Reference profile: In this field, you indicate the profile where the cutoffs for the new profile should be taken from. It is filled automatically if the input table is a result of a site search analysis or derived from it, and it corresponds to the profile used for site search. You also have an option to manually select the reference profile from the available TRANSFAC® profiles.
Cutoffs column: In this field you specify which column of the input table should be considered for the cutoff values. It is filled automatically if the input table is the result of a site search optimization analysis. It is advisable to use “(none)” to leave the cutoffs as in the reference profile.
Output profile: Specify the path to store the result and indicate the name for the new profile.
To launch the analysis, fill the input fields and press [Run].<!– The process will start as shown below:
 –>
–>
After completion of the analysis the output profile is opened automatically as shown below:
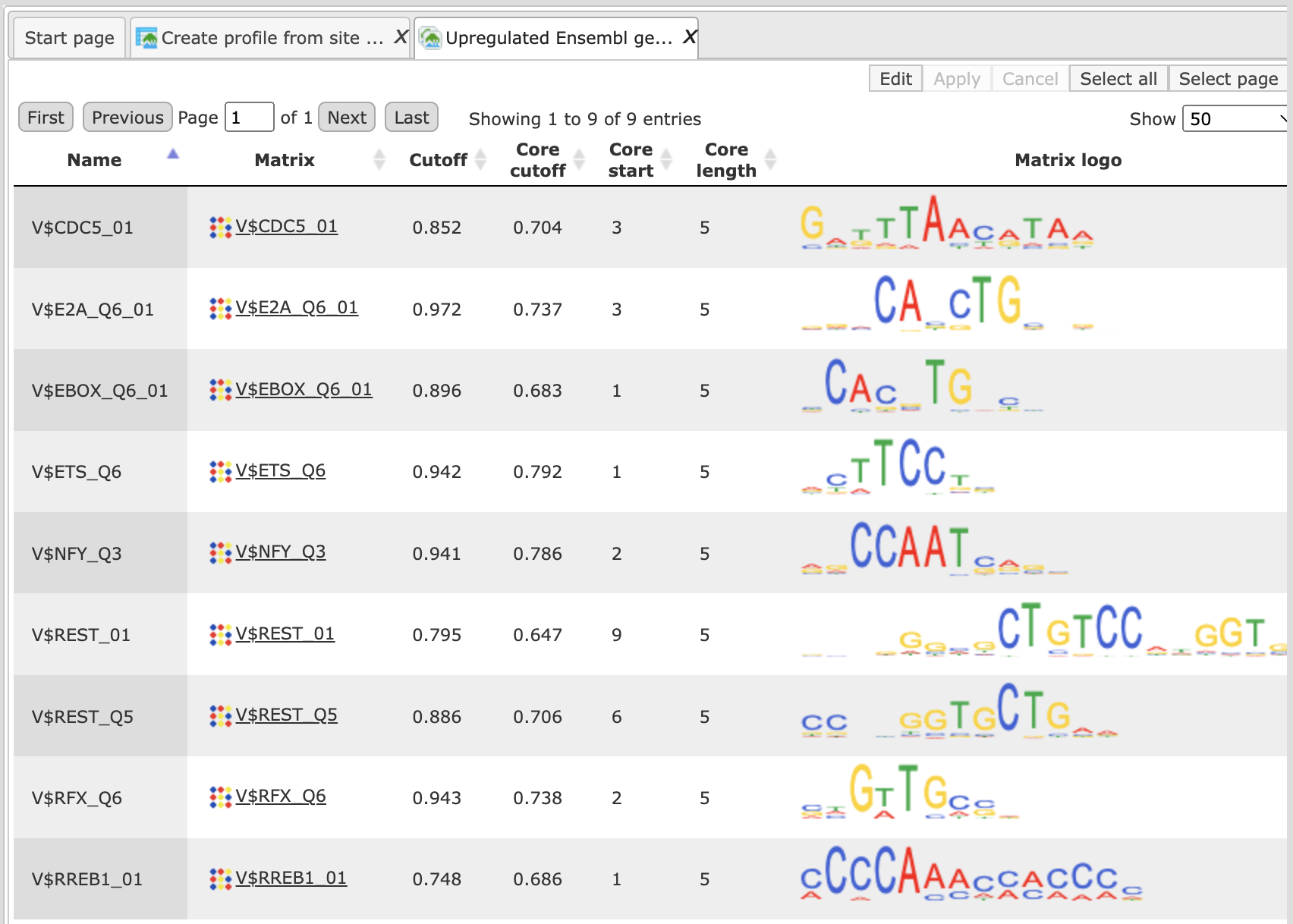
Each row summarizes the information for one site model. In the column Name the name of the site model is given, which in the majority of cases is the same as for the matrix. In the column Matrix name the name for the positional weight matrix is present. For each site model, cutoff is shown in the column Cutoff. According to the TRANSFAC® standard, the core part is specified for each matrix. The core is represented by the 5 consecutive most conserved nucleotides. The columns Core cutoff, Core start and Core length give details about the core of each matrix. In the last column the matrix logo of each matrix is shown.
The new created profile has the symbol , the same symbol as for all other profiles, and is ready to use for the analysis of regulatory regions.
Suggestion when to use “Create profile from table”
Creating a new profile from a table of site models (matrices) might be very useful e.g. if you plan to focus the analysis on those matrices shown to be overrepresented upon the first run of promoter analysis and plan further to construct composite promoter models.
For this, you first run a site search with optimization, e.g. the workflow “Analyze promoters (TRANSFAC®)” or “Upstream analysis (TRANSFAC® and TRANSPATH®)”. Then, you can use the table summary as an input to construct your specific profile.
It will contain only those site models (matrices) for which hits are over-represented in your genes of interest versus the background set, which is a specific subset of the TRANSFAC® matrix library. Having such a profile specific for your genes of interest, you can run it on the same set of genes without optimization, and then use the results as input to construct composite promoter models.
Create profile from matrix library
This tool can set score cutoffs for an entire matrix library and store a corresponding profile, which in turn can be applied binding site analyses.
The figure below shows the default input mask. There are two general ways of setting score cutoffs, either by P-value (the default) or as one cutoff value that will be applied to all matrices.

The input parameters are as follows.
Input matrix library: Selection of the matrix library. All matrices of the library will be present in the profile (“Output profile”).
Core-cutoff: The cutoff for the matrix core, which is the five consecutive matrix position which are most selective for certain nucleotides. When the matrix cutoff is defined by a P-value, the core cutoff will be 0 for all matrices.
Template for cutoffs: The P-value threshold according to which the score cutoff for each matrix will be set. Selecting the “Custom…” option presents a new field to set one common cutoff for all matrices of the library.
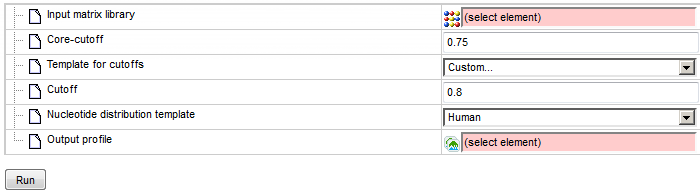
Nucleotide distribution template: The distribution of nucleotides on which P-value calculations are based. Selecting the “Custom…” option enables custom setting of individual base frequencies.
Output profile: The path for the output profile.
An profile can be created as follows.
Step 1. Input the matrix library. As usual, you can drag-and-drop. Use the latest TRANSFAC® library:
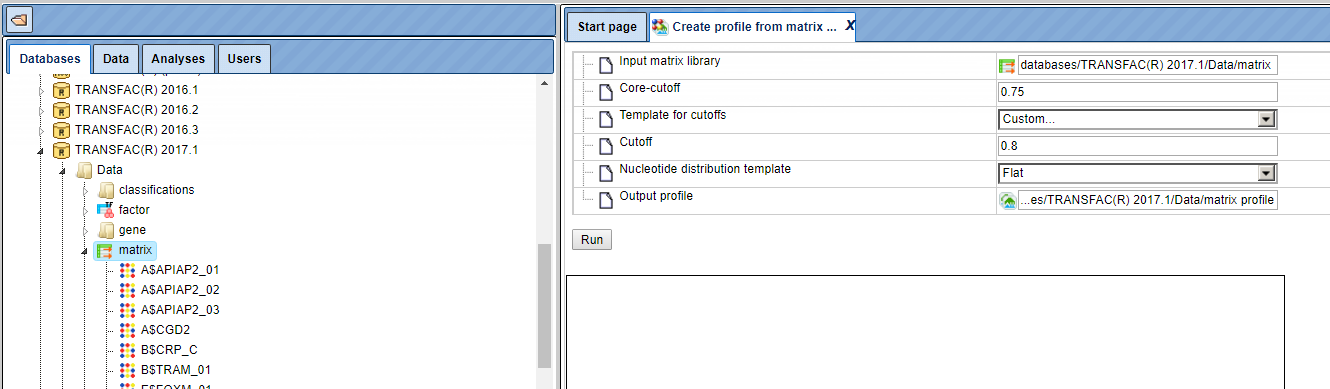
Step 2. Edit other input parameters as shown in the figure above. The output profile path needs to be chosen in a writable directory, e.g. one of your projects.
Search for enriched TFBSs
The platform comprises tools that are dedicated to discovering types of binding sites enriched in a set of sequences. They are named “Search for enriched TFBSs (genes)” and “Search for enriched TFBSs (tracks)”. Both apply a common core algorithm to gene promoters or tracks, respectively. The “Search for enriched TFBSs”-tools are accessible in the “Analysis” tab under “analyses/Methods/Site Analysis”.
Dedication to discovering enriched types of binding sites means that these methods won’t bother with storing predicted binding sites and they also do not require predictions as input. Their sole output is a relatively small table summarizing the enrichment detected for PWM models in a sequence set of interest. As a result, the “Search for enriched TFBSs”-tools are often considerably faster than solutions that have to read and write binding site information. In addition, their outputs consume only the disk space needed to present the results of analyzing binding site enrichment. Hence, a “Search for enriched TFBSs” is for you if you wish to gain insight into enriched binding sites quickly and in an uncomplicated way.
Search for enriched TFBSs (genes)
Let us begin with the analysis for gene promoters. The figure below depicts the input mask of the analysis tool. The parameters are similar to those used by “Site search on gene set” and are described in the following.
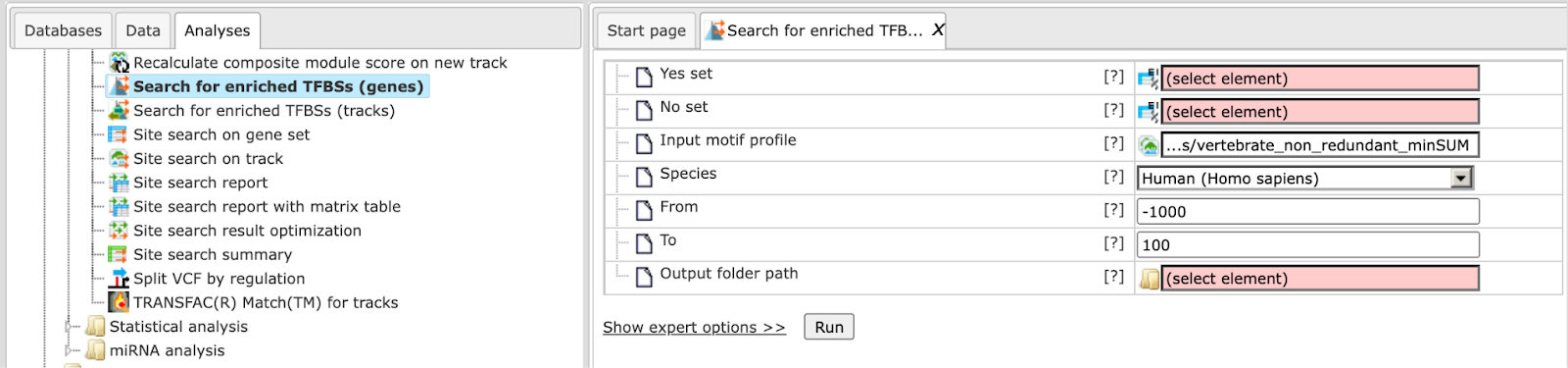
Yes set: This is the set of genes that you want to analyze, for example these can be genes with altered expression. The program accepts genes specified by Ensembl gene identifiers. Note that the “Convert table” functionality in the “Data manipulation” folder can map other identifiers to the required Ensembl genes.
No set: This is the set of background genes (control set), which also need to be specified by Ensembl gene IDs.
These two datasets might be taken from the output tables of previous analyses; see, e.g., “Detect differentially expressed genes”
Species: This option specifies the biological species of Yes and No gene sets. Currently, the method is applicable to human, mouse or rat genes.
From and To: These values define the length and location of promoter regions (upstream and downstream) relative to the transcription start site (TSS). Respective sequence regions are extracted for each Yes and No gene according to Ensembl annotation.
Input motif profile: The profile lists the PWMs (motifs) to be used for binding site prediction together with a score cutoff. By default, this field is set to the last profiled set in your workspace. Note that cutoffs in the profile are ignored, because the “Search for enriched TFBSs” determines a starting threshold specified in the Initial cutoff field.
Output path: In this field you select a path in the workspace to store the output table.
Initial cutoff: This cutoff determines the starting point of the analysis with respect to the score threshold to predict binding sites. The choice is expressed as frequency of predicted sites per base. By default, the analysis begins with a frequency of 5 sites in 100 bases (0.05). From the initial cutoff the algorithm iterates over higher cutoffs and eventually reports the one that resulted in optimal enrichment of binding sites in the Yes set. This is done separately for each PWM.
The steps of an analysis can be described as follows:
Step 1. Input Yes set from the tree. As usual, you can drag-and-drop.<!– Here, the below set of genes are used:
Step 2. Input No set (drag-and-drop). Our example uses the set of “Non-changed Ensembl genes”
Step 3. Select the TRANSFAC® or GTRD profile from the available profiles. In this example, we select the TRANSFAC® profile named “vertebrates_non_redundant_minSUM”:
Step 4. Edit the output path (highlighted green in the figure above). After setting the Yes gene set, a default output path is suggested. The Example folder may however not be writable for your account requiring selection of an alternative such as one of your own projects. A different selection can be made easily by clicking on the field.
We keep the defaults for promoter range (starting from 1000th base upstream to the 100th base downstream of the TSS) and initial cutoff.
Clicking the [Run] button will invoke the analysis.<!– The summary table
is automatically opened in a new tab when the analysis is completed. Here is a part of the output from our example:
 –>
–>
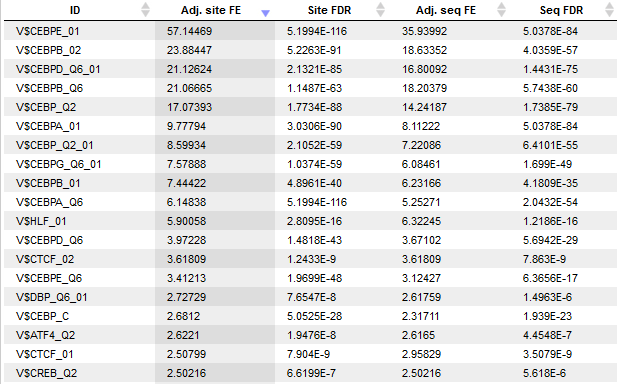
Each row of the output table represents the result for one PWM from the input profile. Fold enrichment (FE) values quantify the enrichment of binding sites in the Yes sequence set as a whole (Adj. site FE) and of Yes sequences with at least one binding site (Adj. seq FE). The FE is an odds ratio that compares the ratio of Yes sites to No sites with the ratio of Yes sequences to No sequences. Unlike in other tools the FE values are statistically corrected (adjusted) quantities that report a value below the actually observed ratio according to the 99% confidence interval. This correction takes into account the underlying site numbers and sequence numbers and penalizes enrichment values that are based on only few binding site occurrences, e.g. at a high score threshold. The output table is sorted by the adjusted Site FE by default.
The statistical significance of enrichment is further assessed by one-tailed binomial test and Fisher tests P-values, for which False Discovery Rates (FDRs) are reported. The Site FDR is based on binomial test P-values calculated for the number of binding site in Yes and No set. The Seq FDR column contains FDRs for Fisher test P-values for the number of sequences with at one site in Yes and No sets.
Additional columns are available via the “Columns” tab of the lower-right panel. Each column is also accompanied by a concise description and can be included into the table presentation upon demand.
Search for enriched TFBSs (tracks)
The “tracks” variant of the “Search for enriched TFBSs” can be applied to
sequence tracks signified by the symbol ( ). For instance, a track may contain genomic intervals identified by a ChIP-seq experiment.
). For instance, a track may contain genomic intervals identified by a ChIP-seq experiment.
The analysis uses the following parameters:
Yes set: This is the track that you want to analyze, for example these can be ChIP-seq intervals bound to a transcription factor.
No set: This is the set of background intervals (control set).
Sequence source: Both Yes and No track need to refer to a common source, such as a genome, as specified by this parameter. Note that you can apply a custom source, e.g. a specifically uploaded genome. Clicking on the “Custom” option will open a new field to choose the custom sequence source.
Input motif profile: The profile lists the PWMs (motifs) to be used for binding site prediction together with a score cutoff. By default, this field is set to the last profiled set in your workspace. Note that cutoffs in the profile are ignored, because the “Search for enriched TFBSs” determines a starting threshold specified in the Initial cutoff field.
Output path: In this field you select a path in the workspace to store the output table.
Initial cutoff: This cutoff determines the starting point of the analysis with respect to the score threshold to predict binding sites. The choice is expressed as frequency of predicted sites per base. By default, the analysis begins with a frequency of 5 sites in 100 bases (0.05). From the initial cutoff the algorithm iterates over higher cutoffs and eventually reports the one that resulted in optimal enrichment of binding sites in the Yes set. This is done separately for each PWM.
The platform provides an out-of-the-box example for this tool under “data/Examples/ Encode TFBS CEBPB in H1-hESC cells”. The ChIP-seq experiment targeted CEBPB binding sites in H1-hESC cells. The steps of an analysis can be described as follows:
Step 1. Input the Yes set from the tree. As usual, you can drag-and-drop. The YES set contains the 500 most significant peaks:
Step 2. Input the No set (drag-and-drop). The NO set contains 1000 random intervals from promoter regions with the same length distribution as the 500 YES intervals:
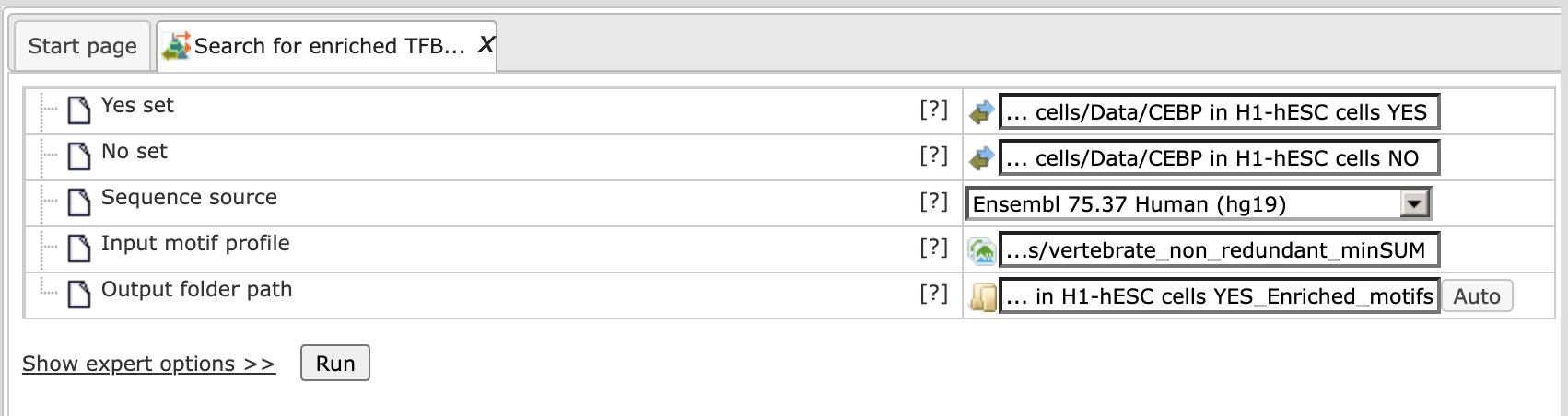
Step 3.
Select the TRANSFAC® or GTRD profile from the available profiles. In this example, we select the latest TRANSFAC® profile named “vertebrates_non_redundant_minSUM”:
Step 4. Edit the output path. After setting the Yes gene set, a default output path is suggested. The Example folder may however not be writable for your account requiring selection of an alternative such as one of your own projects. A different selection can be made easily by clicking on the field.
Clicking the [Run] button will invoke the analysis. The summary table is automatically opened in a new tab when the analysis is completed. A part of the output for our example is shown below. Please refer to “Search for enriched TFBSs (genes)” for further description.
Construct composite modules
Composite modules are combinations of several TFBSs that are found together in a set of regulatory sequences. We search for such combinations of TF binding sites that are overrepresented in the regulatory sequences under study compared to a background set of sequences. The search for composite modules is performed using an in-house implementation of a genetic algorithm. As input for the genetic algorithm we take the output of a site search analysis.
There are two individual analysis functions available with the same symbol  ; they are different with respect to the type of sequences where the search for composite modules is done, and correspondingly with respect to the format of the input data.
; they are different with respect to the type of sequences where the search for composite modules is done, and correspondingly with respect to the format of the input data.
Construct composite modules analysis works on the promoter sequences specified relative to TSS in the set of genes. As input, it takes the results of the Site search on gene set analysis function.
Construct composite modules on tracks works with any DNA sequences specified by their absolute genomic positions, and is very often applied for the analysis of ChIP-seq fragments. As input, it takes the results of the Site search on track analysis function.
Both analysis functions can be found in the geneXplain platform online under the path
https://platform.genexplain.com/bioumlweb/#de=analyses/Methods/Site%20analysis/
Construct composite modules
This analysis function enables the identification of combinations of several TFBSs in the promoters of the genes under study (Yes-set). The resulting composite module differentiates the Yes-set from a background set (No-set).
Before starting this analysis, you need to perform Site search on gene set with your selected Yes-set, No-set and a specified profile of matrices. If you are interested in finding site models for particular TFs, and see them eventually in the resulting composite modules, you need to be sure that such matrices are present in the selected profile. You can use one of the available TRANSFAC® profiles, or alternatively you can construct a customized profile as described earlier. The input form for this analysis is shown below:
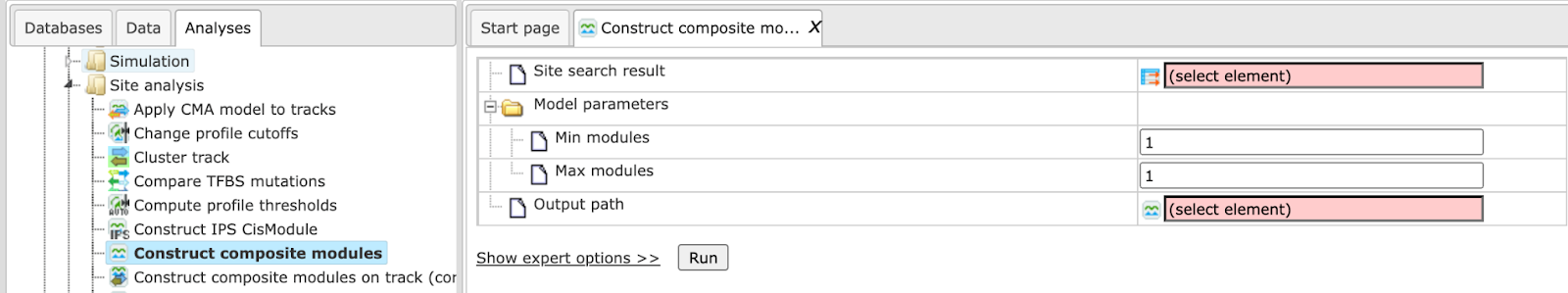
Step 1. Specify Site search result. This is the input field for the
analysis, where you specify the results of the site search. The input field is
marked by the symbol  , which means that the input data set should have the same symbol.
, which means that the input data set should have the same symbol.
As you can see, this is a folder with the results of the Site search on gene set. If you single-click on this folder in the tree area it will be highlighted in blue, and in the Info box you can find a description of all the details about Yes-set, No-set, profile, promoter regions applied to get these results (cf. screenshot below).
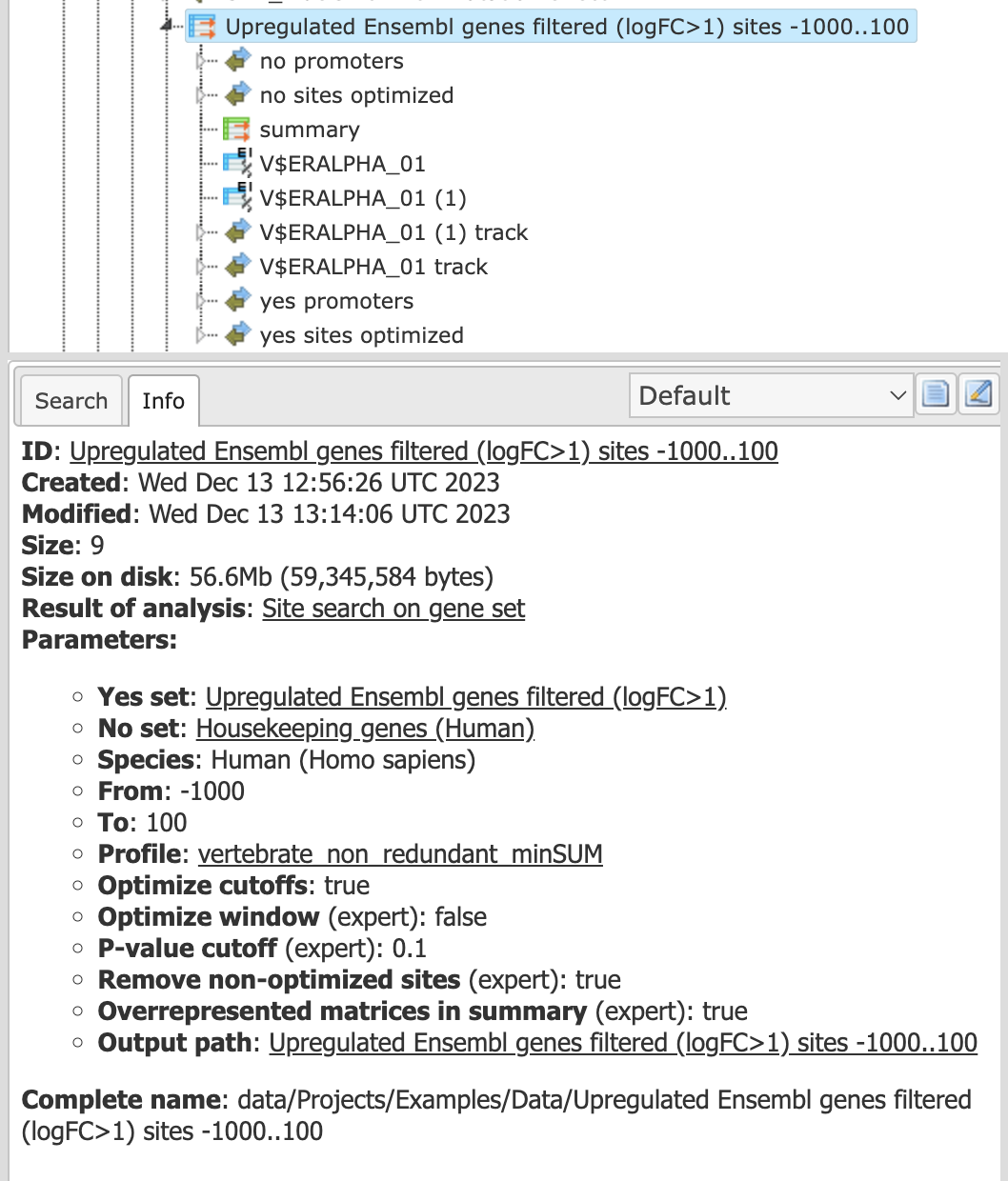
You can drag & drop the name of the folder Upregulated Ensembl genes LogFoldChange >1.2 sites -500..100, profile 0.001 into the input field, or you can select it in the tree in the pop-up window if you click on the pink box of the input field.
Step 2. Specify Model parameters. You can specify the number of elements in the hierarchical structure of the desired composite module. Details and explanations on how to do this are explained in the section Hierarchical structure of the composite modules.
Step 3. Specify Output path. Specify a location for the results in your
project in the tree area. The resulting folder will be marked by the same icon
as the analysis,
Results are described below.
Construct composite modules on tracks
This analysis is designed for identifying combinations of several TFBSs in DNA sequences specified by their genomic positions (tracks). An example of a track that is very often used is a set of the ChIP-seq data. The resulting composite module differentiates between a Yes-track and a background (No-track).
Before starting this analysis, you need to perform Site search on track with your selected Yes-set, No-set and specified profile of matrices. If you are interested in seeing the site models for particular TFs in the resulting composite modules, you have to make sure that such matrices are present in the selected profile. You can use one of the available TRANSFAC® profiles, or alternatively you can construct a customized profile.
The input form for this analysis is shown below:
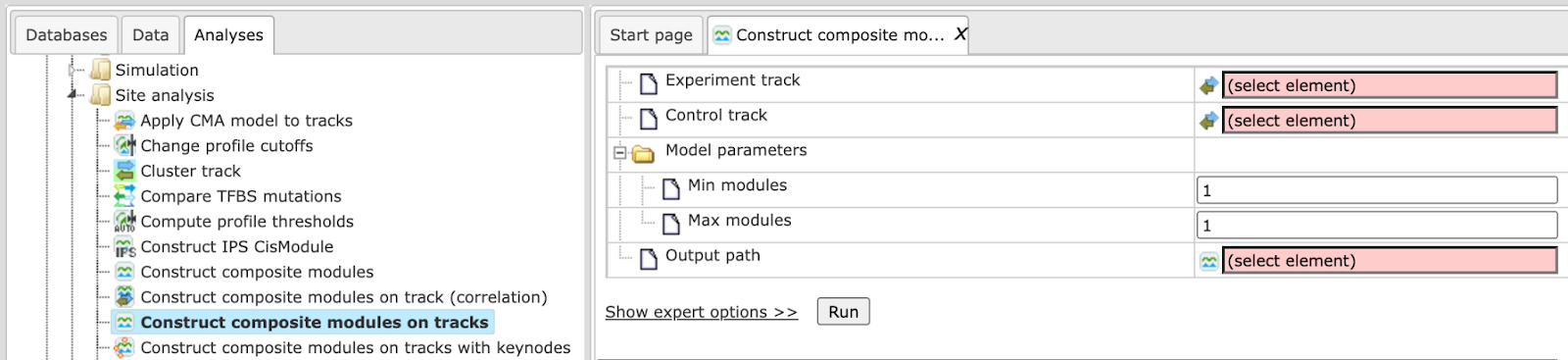
Step 1. Experiment track is the input field for the Yes-track, or track under study.
Step 2. Control track is the input field for the No-track, or background track.
Step 3. Model parameters. You can specify the number of elements in the hierarchical structure of the desired composite module. detailed Explanation can be found at “Hierarchical structure of the composite modules”.
Step 4. Output path. Specify a location for the results in your project in
the tree area. The resulting folder will be marked by the same icon as the
analysis:
Results are described below .
Note. This analysis is the central part of the workflow ChIP-Seq - Identify composite modules on peaks (TRANSFAC®), and it might be more convenient to use the workflow instead of the individual analysis.
Hierarchical structure of the composite modules
Composite modules may have a complex hierarchical structure consisting of two levels: site models and modules. The highest hierarchical level contains several modules and corresponds to the promoter model.
The first level, site model, corresponds to the individual site model, often based on one PWM. Names of the site models are often the same as the matrix names (in case the site models are based on a library of matrices). The site models are taken from the profile that was used in the site search. In the resulting schemas the site models are shown by blue boxes, for instance:
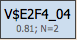
Within these boxes, there are two values below the site model. The first value is the threshold value for the score of the respective site model, which is determined by the genetic algorithm during the optimization process (here it is equal to 0.81); in some cases this value is equal to 0.0, which means that the original threshold value given in the profile was found by the algorithm to be the optimal one. The second value, in this example N=2, is the maximum number of best found individual matches (sites) for this site model which are taken into account for calculating the score of the module.
The next level, module, may contain several site models, shown within the light brown boxes:
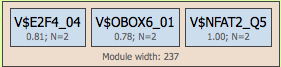
The module is characterized by its width, the average length of DNA window containing matches for the mentioned site models. In the example, the module width is 237 bp. In the resulting schemas modules are shown in green boxes, and they are numbered, e.g. Module 1, Module 2, ….
In the input form you can define the complexity of the promoter model to be constructed by specifying the number of units of each level: number of modules, number of site models, and also the minimum and maximum numbers of individual sites to be considered. In order to illustrate how to specify these parameters, let’s consider three examples of resulting modules depending on the input parameters.
Example 1:
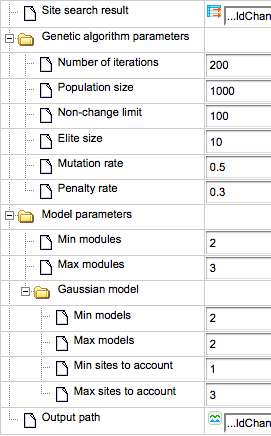
In the picture below (left part) you can see the composite module resulting from the performed analysis. On the right side of the picture the input form with specified parameters is shown.
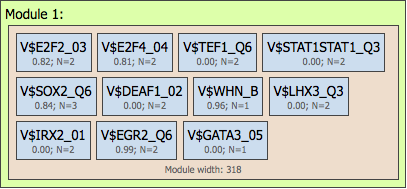
We can see that Min modules (minimum number of modules) and Max modules (maximum number of modules) is 1, and correspondingly there is just one module in the resulting picture, Module 1, highlighted by the red circle on both the resulting schema and the input parameters.
The blue circle highlights the parameters Min models (minimum number of site models within one module) and Max models (maximum number of site models within one module) in the input form. As we can see, the number of site models is set to vary from 4 to 12. Correspondingly, in the resulting schema (left part above) there are 11 site models (blue boxes) identified by the algorithm.
The third parameter, Min sites to account (minimum number of individual sites for each site model to be considered) and Max sites to account (maximum number of individual sites for each site model to be considered), is highlighted by a green circle. As we can see, this parameter is set to vary from 1 to 3, and correspondingly in the resulting schema, for the different matrices we can see N=1 or N=2 or N=3.
Example 2
In this example, the number of modules (red circles) is specified from 2 to 3, and correspondingly the resulting promoter model contains three modules, Module 1, Module 2, and Module 3 (picture below, left part, red circles).
The number of site models is specified in the input form as from 2 to 2, which means that we are going to search for pairs of individual site models. In agreement with the input parameters, in the resulting schema we can see each module containing two site models highlighted by blue circles.
Example 3
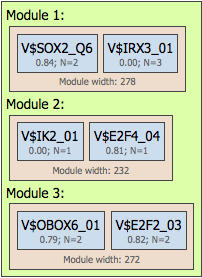
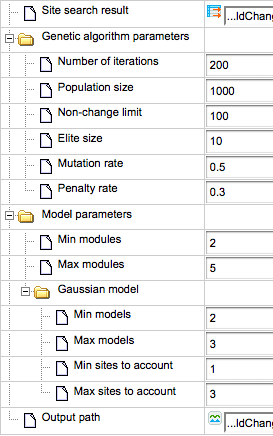
In this example, the number of modules (red circles) is specified from 2 to 5, and correspondingly the resulting promoter model consists of four modules selected by the algorithm, Module 1, Module 2, Module 3 and Module 4 (picture below, left part, red circles).
The number of site models is specified in the input form as from 2 to 3, which means that we would like to find either pairs or triplets of individual site models. In agreement with the input parameters, three out of four modules contain three site models (highlighted by blue circles within Module 1), and one module, Module 3, contains two site models.

Based on these three examples you can specify input parameters for the number of modules, site models, and individual sites, depending on what resulting promoter model you would like to get.
Site models in focus
There are situations when researchers would like to focus on particular TFs and would like to find out with what other TFs they may form composite modules. Site models that must be present in the resulting modules, are referred to as site models in focus. In the expert options menu, under Score calculation parameters, there is a field Site models in focus. As soon as the site search results are submitted to the input field, all site models from the profile used before for site search are now available for selection via the drop-down menu, as shown on the screenshot below.

In this example, we can see that V$AP1_Q2_01 is selected. Using the Ctrl button, several matrices from the list can be selected.
As soon as the site model is selected, two new fields will appear just below the field Site models in focus (screenshot below).
All modules contain site model in focus. When this box is checked, all the resulting modules must contain specified site models. When it is unchecked, at least one of the resulting modules must have a specified site model, and other may have them too, but not necessarily.
Focused sequences percent. In the drop-down menu, you can specify the minimum percentage of the Yes sequences (genes, promoters) that must contain modules with the specified site models in focus.

Visualization and interpretation of the results
Let us consider the results of the Construct composite module analysis obtained for the following input data set.
Input parameters used were the following:

As result, a new folder is generated containing two tables, two tracks, and one histogram, as shown below.

Model visualization in Yes set
This table represents the primary results of the analysis, and shows the visualization of the identified composite modules in the promoters of the Yes set.

Each row in this table corresponds to one gene of the Yes set, and for each gene the Ensembl ID and the gene symbol are shown in the two first columns. The column Model displays a symbolic map of the gene promoter taken for the analysis, in this case -500/+100 relative to the TSS. Arrows of different colors correspond to individual TFBSs, and a gradient in grey corresponds to the statistical density of the identified composite modules. The most intensive grey color corresponds to the center of a composite module. Each individual TFBS on this map is clickable, and upon a click information is displayed in the Info box (bottom left corner in the tool). As an example, one blue arrow is selected on the promoter of the top gene in the screenshot above, and for this selected TFBS the following details are shown in Info box:

The last column in the table, Score, shows a score calculated for each promoter depending on the number of modules, site models, sites, their scores and other statistical parameters. The higher the score for a promoter, the better the differentiation of this promoter from the promoters of the No set. The column Score is used for default sorting of the table, with the highest scores on top.
Having opened the table Model visualization on Yes set, you can see the schematic representation of the hierarchical structure of the identified composite module as well as a comprehensive set of its statistical characteristics at the bottom part of the tool, under My description tab, as shown on the screenshot below.

Yes track
The Yes track provides essential information about the regulation of individual promoters and is therefore important to be included in the visualization of individual promoters by the genome browser.
The schematic visualization can be comfortably extended to a more detailed visualization for each individual promoter.

To do this, open the table Model visualization on Yes set, select one row in the table with a mouse click, and open the tab Genome browser in the Operation Field (bottom right part of the tool),.
For a selected promoter, you can see a more detailed map, including the names of the matrices and the numbers of individual modules, M1 through M4. Each element of this interactive map has a corresponding check box. Unchecked elements will not be displayed on the map. De-selection is applied simultaneously to both the detailed view of one promoter, and the table with the schematic representation of all promoters.
To adjust colors for individual matrices, you can open the next tab to the right, called Site colors, and change the colors to your liking by clicking on the individual colored box, as shown below.
Model visualization on No set and No track
The table Model visualization on No set shows a visualization of the identified composite modules in the promoters of the No set.
The structure of this table is the same as that of the Model visualization on Yes set table, described above.
The function of the No track is to provide a possibility for a detailed visualization of no promoters in a way similar to that of the Yes track.
Histogram
The distribution of scores for individual promoters is shown as a histogram, where the promoter score value is shown on X axis and the percentage of promoters (% sequences) having this score is shown on the Y axis.
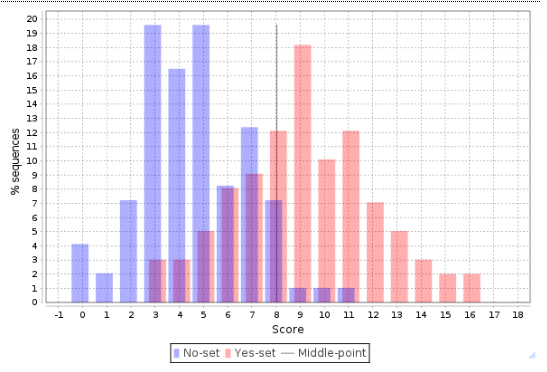
This histogram can be further interpreted applying the statistical characteristics described above.
The center, a vertical grey line, corresponds to the average score value and is equal to 3.44 in this example. Promoters from the No set with a score above 3.44 are shown in the histogram as blue bars to the right of the center, and they are referred to as false positives. In this example, the false positive rate is 16.82 %.
Promoters from the Yes set with a score below 3.44 are shown in the histogram as red bars to the left of the center, and they are referred to as false negatives. In this example, the false negative rate is 23.42 %.
A visual analysis of the histogram suggests that the Yes promoters with a score above 4.5 are very well separated from the No promoters, which means that for this part of the promoters the composite model constructed is most suitable. In this example there are 38 promoters with the score value >4.5; they can be saved as a separate gene set, and for them the model obtained works best.
Score calculation of the composite models
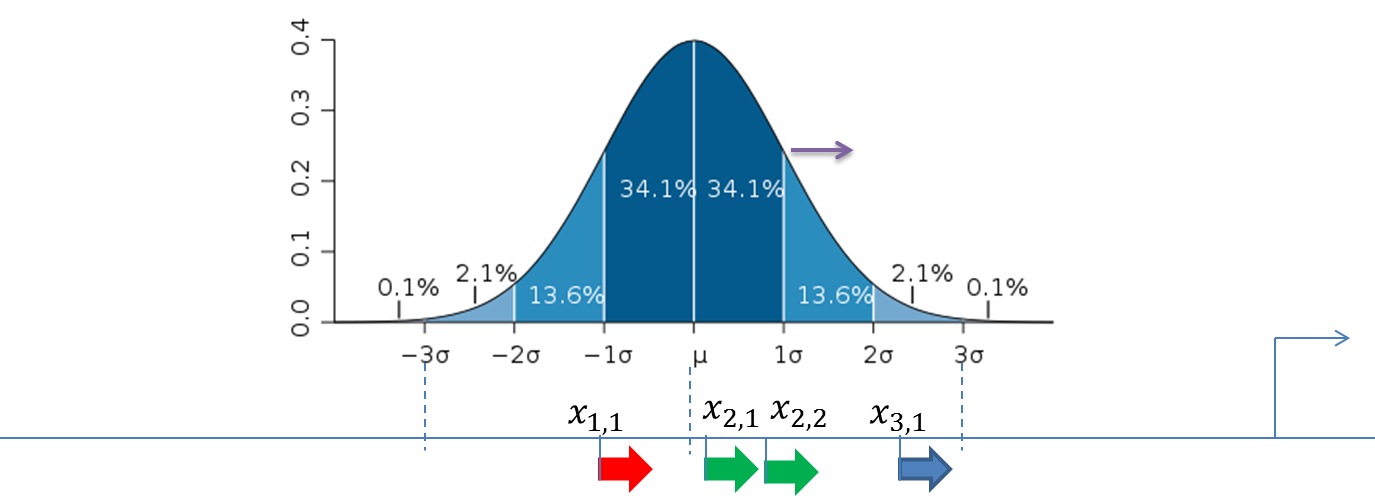
The figure below demonstrates the calculation of the score value for the composite modules in the promoter sequences. The TSS is shown as a thin arrow on the right side of the figure. Four thick arrows exemplify four sites found in this promoter. The color of the arrows exemplifies the site model which these sites belong to (three site models – red, green and blue).
A promoter model consists of K modules. The score of each module Mk (Score(Mk), k = 1, …, K) is calculated according to this formula:
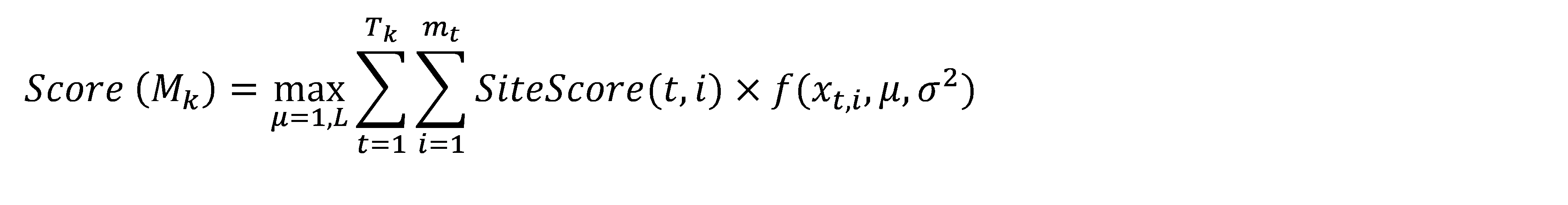
Here, Site Score (t,i) is the site score for the sites found in the promoter, which is calculated by the Match algorithm.
mt – the number of sites of the site model t found in the promoter.
Tk – the number of site models in the module Mk, and
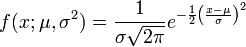
The final promoter score is calculated as the sum of the module scores Mk.
Standard deviation (σ) of the normal distribution is subject of optimization by the genetic algorithm and represents the width of the module in the output of the composite module analysis.
About the GSEA analysis and the interpretation of the results
Schematic description of the GSEA algorithm
The Gene Set Enrichment Analysis (GSEA) is a method that determines whether an independently defined set of genes shows a statistically significant enrichment either in up-regulated or down-regulated genes http://www.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html
Independently defined sets of genes might be groups of genes linked to Gene Ontology terms, or to TRANSPATH® pathways, or to Reactome pathways, etc.
As input for the GSEA, we can use either normalized tables after normalization of the microarray raw data, or a table with genes that contains a column with pre-calculated expression values. The algorithm of the GSEA can be schematically presented in three steps:
All genes are sorted by Fold Change, from highly up-regulated to highly down-regulated. In such a sorted table, each gene is given a rank, which is just the number of the line where this gene is located in the sorted table. The most highly up-regulated gene has the rank “1”, and so on.
The algorithm takes a set of genes linked to one biological term (e.g. to a particular GO category) and maps each gene in this set to the sorted (ranked) table. Genes in some categories might be distributed randomly over the ranked table; some are among the up-regulated, some among the down-regulated, and some among the non-changed genes. For some of the GO categories, a majority of the genes are among the up-regulated. These GO categories are the most interesting for a biological interpretation.
Several statistical parameters, ES (Enrichment Score), NES (Normalized Enrichment Score), Rank at max, nominal p-value, FDR, for each GO category are calculated. ES and NES reflect how significantly genes in each particular GO category are enriched (over-represented) among up-regulated genes.
Structure of the resulting tables with enriched ontological terms
The resulting tables with enriched ontological categories contain twelve columns. Here, to have a better resolution of the screenshots, one table is shown in two parts. This is the result of a GSEA using the HumanPSD™ biological process.
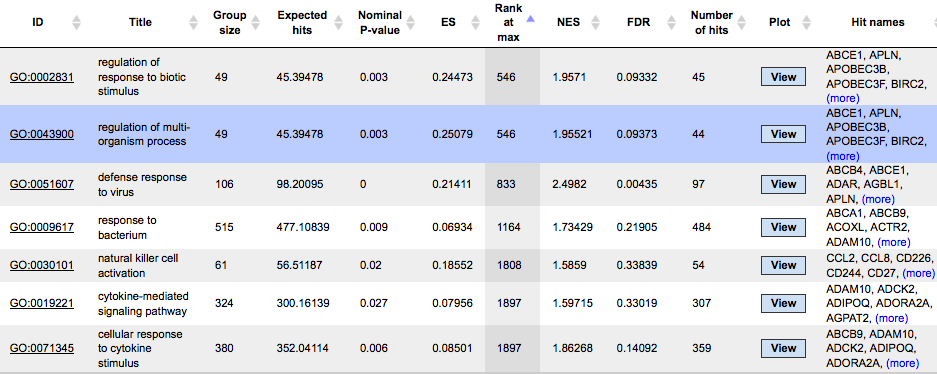
Each row presents details about one enriched ontological term. The column ID comprises the identifier of the ontological category, here identifiers of Gene Ontology biological process terms. These identifiers are hyperlinked to the page http://www.ebi.ac.uk/QuickGO/ where you can get further information about this ontological term. In case of enrichment by HumanPSD™ diseases the disease identifiers are hyperlinked to http://ctdbase.org/
The columns Title and Group size contain further details about the ontological terms, the title and the number of genes linked to this term in the corresponding database, here in HumanPSD™.
The column Expected hits shows the number of genes expected to fall into this category by random chance, based on the size of the input set and the size of the category.
Five columns, Nominal P-value, ES (Enrichment Score), Rank at max, NES (Normalized Enrichment Score), and FDR, show corresponding statistics of the results. For more details about each of these statistics, please refer to http://www.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html, under the section Interpreting GSEA results/GSEA statistics.
The column Number of hits shows how many genes from the input set fall into the enrichment group; these genes are explicitly listed in the column Hit names. As the lists can get quite long, only a few genes are shown by default in each row. To get the full list, press (more). The column Plot and View diagrams are described in detail in a separate subsection below.
Further possible actions with the enrichment results.
The tables with the GSEA results can be exported in txt, csv or html formats. To export, you can apply the Export button in the top control menu, highlighted by the dark-blue oval.
One or several rows of the table can be selected with shift-click , shown in blue color in the screenshot below. Having selected one or several rows in this way, you can save hits of these rows in a separate gene table, with the button Save hits in the top control menu, highlighted by the red oval. Such genes tables can be analyzed further, e.g. to find master regulatory molecules in the networks, and to identify transcription factors that might commonly regulate these genes.
You can save the selected rows in a separate table, with the button Save selected rows as… in the top control menu, highlighted by the green oval.
Enrichment plots
As described above, there is a column Plot with the buttons View in each row in the GSEA resulting tables.
Let’s open a visualization plot for the category defense response.
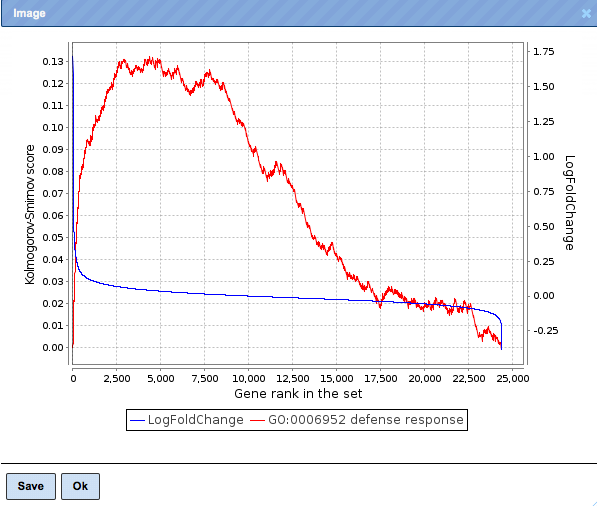
X-axis
On this line, all (human in this example) genes present in the input table are ranked from 1 to approximately 23,000 according to their LogFoldChange. The numbers we see on the X-axis, 2,500; 5,000 etc., are the ranks of the genes.
At the left end of the line X, up-regulated genes are located, and down-regulated genes at the right end . The smaller the rank of a gene is, the higher is its up-regulation.
Y-axis, right
This axis shows the values of the LogFoldChange. Positive values correspond to up-regulated genes, negative values to down-regulated genes.
Blue plot
This is a distribution of LogFoldChange values among all (human) genes.
Y-axis, left
This axis shows the values of the ES parameter (also known in statistics as Kolmogorov-Smirnov score). Positive values correspond to up-regulated genes, negative values to down-regulated genes.
Red plot
This is a distribution of ES values for a particular GO category over all (human) genes. ES is calculated by summing up on the previous values, so the most interesting parts for us are those where the plot is coming up.
Interpretation of the plots
For interpretation, let’s look at the red plot. The most interesting parts are those where the red line is ascending. In this example plot, let’s follow the red line from its left end to its maximum.
The starting point for the red line is 0 on the Kolmogorov-Smirnov score axis.
Next, the algorithm takes each gene within the category “defense response”, and
looks at what the LogFoldChange for this gene is. If a gene is up-regulated, the red
line grows. Then the algorithm takes the next gene from “GDP binding” and again
looks at what the LogFoldChange for the second gene is. If it is also up-regulated, the
red line grows further. This is called incremental growth: the closer the next
gene is to the previous one on the X-axis, the more pronounced is the growth of
the red line.
Let’s add an auxiliary line (here, the green dotted line) from the maximum point of the red plot down to the X-axis. The intersection of the green dotted line with X-axis corresponds to the rank of the gene at maximum enrichment. This value is exactly what in statistics is called Rank at max, and it is shown in a dedicated column in the tables with the GSEA results. In the plot shown above, the green line crosses the X-axis at approximately 4,500. To know this value exactly, look at the table, and in the column Rank at max for this category, you can find the number 4328.
To describe this plot, we can say:
“Genes belonging to the GO category defense response are enriched among the top 4328 up-regulated genes”.